 Peng Ouyang1†
Peng Ouyang1† Lijuan Wang2†Jianlong Wu1†Yao Tian1Caiyun Chen1Dengsheng Li1Zengxi Yao1Ruichang Chen1
Lijuan Wang2†Jianlong Wu1†Yao Tian1Caiyun Chen1Dengsheng Li1Zengxi Yao1Ruichang Chen1 Guoan Xiang3*Jin Gong1*Zhen Bao1*
Guoan Xiang3*Jin Gong1*Zhen Bao1*- 1Department of General Surgery, The First Affiliated Hospital of Jinan University, Guangzhou, Guangdong, China
- 2Department of Pathophysiology, School of Medicine, Jinan University, Guangzhou, Guangdong, China
- 3Department of General Surgery, Guangdong Second Provincial General Hospital, Guangzhou, Guangdong, China
Immune Checkpoint Inhibitors (ICIs) therapy has advanced significantly in treating malignant tumors, though most ‘cold’ tumors show no response. This resistance mainly arises from the varied immune evasion mechanisms. Hence, understanding the transformation from ‘cold’ to ‘hot’ tumors is essential in developing effective cancer treatments. Furthermore, tumor immune profiling is critical, requiring a range of diagnostic techniques and biomarkers for evaluation. The success of immunotherapy relies on T cells’ ability to recognize and eliminate tumor cells. In ‘cold’ tumors, the absence of T cell infiltration leads to the ineffectiveness of ICI therapy. Addressing these challenges, especially the impairment in T cell activation and homing, is crucial to enhance ICI therapy’s efficacy. Concurrently, strategies to convert ‘cold’ tumors into ‘hot’ ones, including boosting T cell infiltration and adoptive therapies such as T cell-recruiting bispecific antibodies and Chimeric Antigen Receptor (CAR) T cells, are under extensive exploration. Thus, identifying key factors that impact tumor T cell infiltration is vital for creating effective treatments targeting ‘cold’ tumors.
1 Introduction
In recent years, Immune Checkpoint Inhibitors (ICIs) have increasingly been incorporated into the treatment of various cancers, becoming a standard part of oncological treatment guidelines. However, a significant proportion of cancer patients still exhibit poor responses to ICI therapy. This trend highlights a need for further research and development in personalized cancer treatment strategies to improve outcomes for this patient subset (1, 2). In patients with solid tumors, ‘hot’ tumors (‘immune- inflamed’) often show a favorable response to ICIs, characterized by extensive lymphocyte infiltration in the tumor parenchyma. In contrast, ‘cold’ tumors exhibit a poorer response to ICIs. These tumors are marked by an inability of T cells to penetrate the tumor parenchyma, remaining instead in the tumor stroma (‘immune-excluded’) or by a lack of T cell infiltration in both the tumor parenchyma and stroma (‘immune-desert’) (3). This distinction underscores the importance of understanding tumor immunology to optimize ICIs therapy efficacy. However, increasing evidence suggests that not all tumors with high T cell infiltration exhibit favorable responses to ICIs. Conversely, some tumors with low T cell infiltration may also demonstrate good responsiveness to ICIs. This observation indicates a more complex relationship between T cell infiltration levels and ICI response, underscoring the need for a deeper understanding of tumor immunobiology to effectively predict and enhance ICIs therapy outcomes (4–6). These findings indicate that T cell infiltration might be necessary, but additional factors may be required for precisely identifying the responsiveness to ICIs. Currently, the treatment of ‘cold’ tumors remains a significant challenge. In this review, we discuss the definitions of ‘cold’ and ‘hot’ tumors, as well as the challenges the immune system may encounter at different stages of the cancer immunity cycle. We also describe therapeutic approaches combining ICIs with other strategies to overcome ‘cold’ tumors. This integrative approach aims to enhance the understanding and treatment efficacy of tumors with varying immune characteristics.
2 Definition of “cold” and “hot” tumors
The concept of ‘cold’ and ‘hot’ tumors is not new in the field of oncology. It was first described in 2006 by Galon et al. in their publication on the relationship between immune cell types, density, and distribution with the prognosis of colorectal cancer. This seminal work introduced the idea of classifying tumors as ‘hot’ or ‘cold’ based on the type, density, and distribution of immune cells within the tumor microenvironment. They posited that this immune-based classification in colorectal cancer could provide a more accurate prognosis assessment than the traditional TNM staging system. This approach underlines the significant role of the immune landscape in understanding and predicting cancer progression (7). In 2007, Galon and colleagues proposed the concept of “immune contexture” based on immunoscore (8). Following this, in 2009, Camus et al. first described three immune coordination profiles (hot, altered, and cold) in primary colorectal cancer (CRC), balancing tumor escape and immune coordination (9). Building on these works, researchers introduced the immunoscore, which assesses the infiltration of lymphocyte populations (CD3 and CD8) in the tumor core and at its margin. The score ranges from immunoscore 0 (I0, low-density CD3 and CD8 stained cells in the tumor center and periphery) to immunoscore 4 (I4, high-density CD3 and CD8 stained cells in these regions) (10, 11). This scoring system classifies cancer based on immune infiltration and introduces the concepts of ‘hot’ tumors (I4) and ‘cold’ tumors (I0-I3). As research progressed, the characteristics of hot tumors were expanded to include the presence of tumor-infiltrating lymphocytes (TILs), expression of programmed death-ligand 1 (PD-L1) on tumor-associated immune cells, and a high tumor mutational burden. Conversely, cold tumors, characterized by poor infiltration, also feature low or negligible PD-L1 expression, high proliferation rates, and a low tumor mutational burden (12).
3 Mechanisms of immune escape in “cold” tumors
Immune checkpoints encompass a group of receptors expressed by immune cells, facilitating the dynamic regulation of immune homeostasis. They hold particular relevance for the functioning of T cells. Among these checkpoints, PD-1 and its primary ligand PD-L1 find expression on T cells, tumor cells, and myeloid cells infiltrating tumors. The interaction between PD-1 and PD-L1 leads to CD8+ T cell exhaustion, a potentially irreversible state of dysfunction characterized by diminished or absent effector functions (including cytotoxicity and cytokine production), reduced responsiveness to stimuli, and altered transcriptional and epigenetic profiles (13, 14). Tumor cells exploit this interaction to establish immune tolerance. However, it also serves essential physiological roles, such as limiting autoimmune inflammation, preserving fetal tolerance during pregnancy, and preventing the rejection of transplanted organs (15). Immune checkpoint inhibitors function by blocking immune checkpoints, thus restoring the anti-tumor activity of CD8+ T cells. Immune checkpoint inhibitors function by blocking immune checkpoints, thus restoring the anti-tumor activity of CD8+ T cells. Any failures during the stages of T cell activation, homing, or infiltration into the tumor bed in the tumor immune process can result in inadequate T cell infiltration into the tumor core (Figure 1). This, in turn, leads to resistance to ICIs therapy.
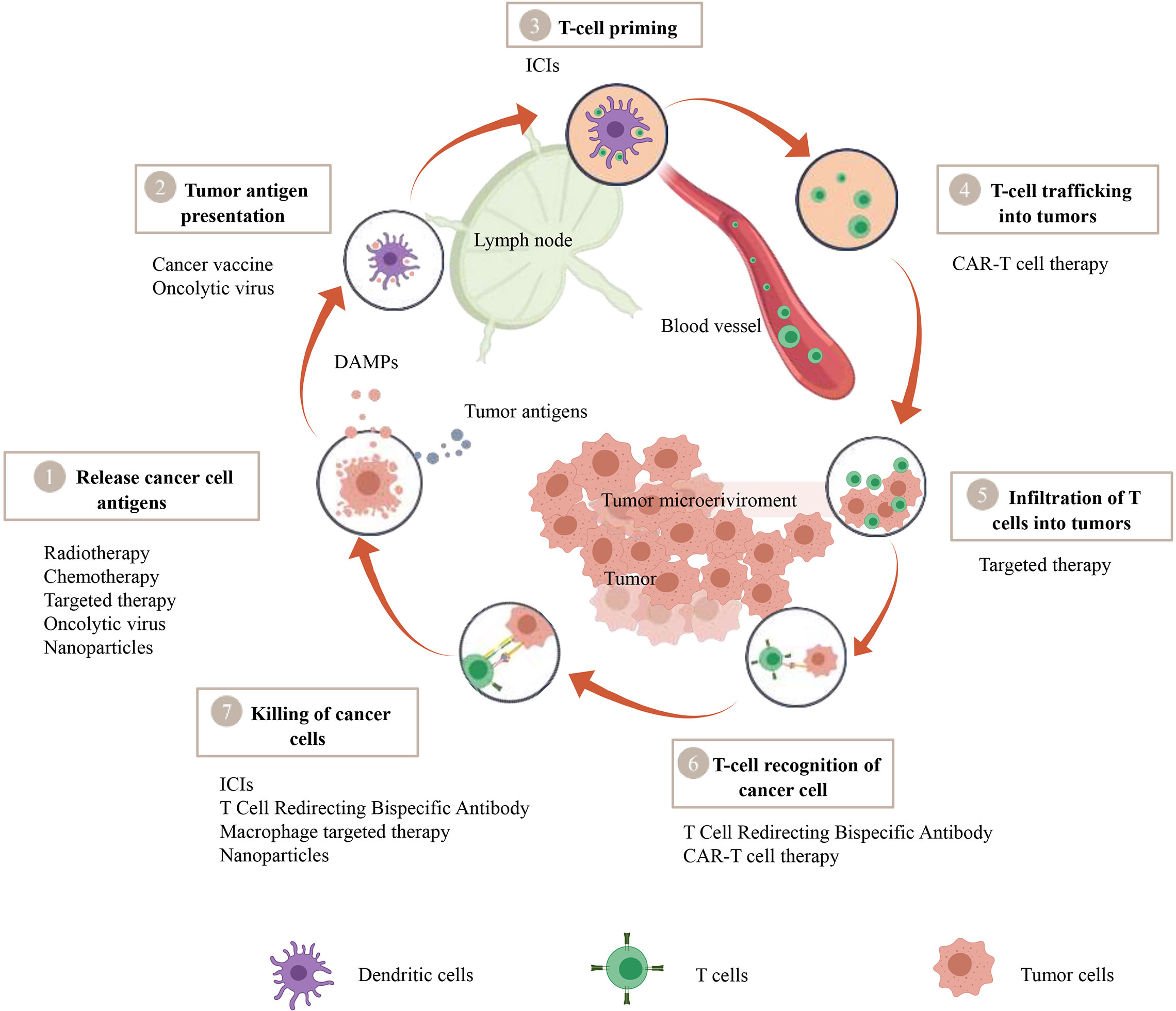
Figure 1 A therapeutic strategy to convert cold tumors into hot tumors based on tumor immune cycle. The cancer-immunity cycle encapsulates seven pivotal steps, with each one being integral to the overall mechanism. A malfunction or inefficiency at any juncture can potentially instigate the tumor to evade the immune response. Nevertheless, a wide array of therapeutic approaches such as Chimeric Antigen Receptor T-cell (CAR-T) therapy, T-cell Redirecting Bispecific Antibodies, cancer vaccines, oncolytic viruses, macrophage-targeted therapies, radiotherapy, chemotherapy, targeted therapies, and nanoparticle-assisted treatments manifest their potential to modulate this cycle, thereby amplifying the body’s defensive reaction against tumors.
3.1 Lack of tumor antigens
Tumor antigens can be categorized into two main types: Tumor-specific antigens (TSAs) and Tumor-associated antigens (TAAs) (16). TAAs are antigens that, while not exclusive to tumor cells, are present in normal cells and other tissues but are significantly elevated during cellular transformation into cancer. These antigens exhibit quantitative changes without strict tumor specificity. Although they can also trigger immune responses, the most crucial in activating immune responses are neoantigens, also known as TSAs. TSAs are antigens unique to tumor cells or present only in certain tumor cells and not in normal cells. This includes antigens produced by oncogenic viruses integrated into the genome and those arising from mutant proteins (17). In addition to mutations in DNA coding regions, gene fusions (18), mutations in non-coding regions (19), and alternative splicing (20) can also generate neoantigens. Loss of DNA damage response can lead to gene mutations, including mismatch repair deficiencies (dMMR) and microsatellite instability (MSI) (21). Currently, ICIs treatment has become the preferred therapy for advanced colorectal cancer with high microsatellite instability (22). Therefore, the recognition of TSAs plays a key role in activating T cells and promoting their infiltration into tumor tissues.
Tumor Mutational Burden (TMB) refers to the number of nonsynonymous single nucleotide mutations found in tumor cells. A high TMB implies more mutations, leading to the generation of more TSAs. Research over the past five years has shown that tumors with high TMB respond better to ICIs treatment than those with low TMB (23). McGrail et al. found that in cancers characterized by recurrent mutations, neoantigens are positively correlated with TILs infiltration (24). However, in tumors characterized by recurrent copy number variations, there is no correlation between TILs infiltration and the neoantigen load (24). Spranger et al. found no association between T cell infiltration and nonsynonymous somatic mutations (NSSMs) (25). These studies indicate that the lack of T cell infiltration cannot be solely explained by TMB.
3.2 Defective antigen presentation
Dendritic cells (DCs) are pivotal in the antigen presentation process. They play a critical role in initiating anti-tumor immune responses by capturing and processing tumor antigens, conducting cross-presentation, and activating naive T cells. There are multiple subgroups of DCs, including classical DCs (type 1 cDC1 and type 2 cDC2), plasmacytoid DCs (pDCs), inflammatory DCs, and Langerhans cells. Each subgroup plays a distinct role in immune responses (26). In tumor immunology, DCs are often activated by “danger signals” such as Damage-Associated Molecular Patterns (DAMPs), including ATP, HMGB1, Calreticulin (CRT), and the S100 protein family (27). Among the different DCs subtypes, cDC1s are particularly crucial in tumor immunity. Studies show that Batf3-knockout mice, which lack cDC1s, exhibit reduced TILs and decreased responsiveness to ICIs (28). Research on tumor-bearing mice indicates that cDC1s are essential for reactivating circulating memory anti-tumor T cells and responding to ICIs (29). Tumors can evade detection by DCs through various mechanisms, such as expressing the “don’t eat me” signal CD47 (30). Tumor cells also avoid exposing DAMPs, such as CRT, by expressing inflammatory molecules like A20 (31) in CRC, STC1 (32) in certain tumors, and glycosylated B7-H4 (33) in breast cancer. cDC1s cross-present antigens from dying tumor cells, which is fundamental in initiating anti-cancer CD8+ T cell responses. cDC1s express high levels of DNGR-1 (also known as CLEC9A), a receptor that binds to exposed F-actin in dying tumor cells and facilitates antigen cross-presentation. Tumor cells can inhibit this process by secreting extracellular proteins like sGSN, reducing the binding between DNGR-1 and F-actin, thus preventing cDC1s from activating CD8+ T cells (34).
HLA-I Loss of Heterozygosity (HLA-I LOH) is a significant mechanism of immune escape, with approximately 17% of tumors exhibiting HLA-I LOH (35, 36). TRAF3, a factor that inhibits NF-κB activity, negatively regulates the expression of MHC-I. Lower levels of TRAF3 are associated with better responses to ICIs) (37). Notably, MHC-I on the surface of Pancreatic Ductal Adenocarcinoma (PDAC) cells is degraded through autophagy. Inhibiting autophagy can restore MHC-I levels on the surface of PDAC cells. In mouse models of PDAC, combining autophagy inhibitors with dual ICIs enhances the immune response against the tumor (38). Therefore, increasing the expression of HLA-I on tumor cells’ surface could be a potential strategy for treating ‘cold’ tumors.
3.3 T lymphocytes are unable to infiltrate the tumor bed through the blood circulation
3.3.1 Dysregulation of chemokines and cytokines
Chemokines in the TME mediate the recruitment of various immune cells, including T cells, thereby influencing tumor immunity and treatment outcomes. Dysregulation of chemokines within the TME often promotes tumor progression by altering the infiltration of immune cells. For instance, effector CD8+ T cells, Th1 cells, and NK cells can migrate into the tumor in response to chemokines like CXCL9 and CXCL10, facilitated by their shared expression of the CXC chemokine receptor 3 (CXCR3) (39). Enhancer of zeste homologue 2 (EZH2) and DNA methyltransferase 1 (DNMT1) reduce the presence of effector T cells in tumors by inhibiting the production of CXCL9 and CXCL10 by Th1 cells (40). In colorectal cancer, the polycomb repressive complex 2 (PRC2) similarly suppresses the production of these chemokines by Th1 cells, thereby diminishing the entry of effector T cells into the tumor (41). Additionally, the expression of CCL5 is associated with the infiltration of CD8+ T cells, while DNA methylation leads to reduced expression of CCL5, consequently decreasing TILs (42). Reactive nitrogen species (RNS) produced in the TME can also induce nitration of CCL2, impeding T cell infiltration (43).
Cytokines significantly impact tumor cell development and the treatment outcomes of ICIs. For instance, in urothelial cancer, combining Transforming Growth Factor β (TGF-β) blockade with ICIs therapy has been shown to promote T cell infiltration into the tumor core and elicit strong anti-tumor immune responses (44). Similarly, in colorectal cancer, inhibiting TGF-β increases the number of cytotoxic T cells, thereby inhibiting tumor metastasis (45). Additionally, Interferon γ (IFNγ), Interleukin-2 (IL-2), and Interleukin-9 (IL-9) also play crucial roles in the efficacy of ICIs treatment (46).
3.3.2 Immune cell–mediated immunosuppression
Within the tumor microenvironment, tumor cells interact with various immune cells that have immunosuppressive functions, particularly regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs), playing a crucial role in the regulation of tumor development and progression (47, 48).
Tregs, initially identified as thymus-derived immunosuppressive cells among CD4+ T cells with a high expression of CD25 in mice (49), and later described in humans (50–52), gained recognition in the field of immunology. The discovery of Foxp3, a master regulator of Tregs, firmly established this population as an independent immunosuppressive cell lineage within CD4+ T cells (53–55). In current classification, Tregs are divided into natural/thymic and peripherally induced subsets, based on the sites of their development (56–58). Hence, it becomes imperative to distinguish Tregs from FOXP3-expressing conventional T cells in humans. In human studies, FOXP3-expressing CD4+ T cells are further categorized into three groups, depending on the expression of CD4, CD45RA, CD25, and/or FOXP3: 1) naive/resting Tregs, defined by CD4+CD45RA+CD25lowFOXP3low T cells; 2) effector/activated Treg (eTreg) cells, characterized by CD4+CD45RA−CD25highFOXP3high T cells; and 3) non-Treg cells, identified as CD4+CD45RA−CD25lowFOXP3low T cells. Naive Tregs, initially displaying weak suppressive activity, have recently exited the thymus but remain quiescent in the periphery (59, 60). Upon TCR stimulation, naive Tregs exhibit vigorous proliferation and differentiate into highly suppressive eTreg cells. In contrast, non-Treg cells lack immunosuppressive functions and instead produce inflammatory cytokines, including interferon (IFN)-γ and IL-17 (61). Treg cells play a crucial role in dampening antitumor immune responses, particularly those directed towards tumor-specific effector T cells (62). These Treg cells are attracted to the TME, where they undergo local proliferation and differentiation into an activated subset with potent suppressive capabilities (63). Importantly, the presence of a high frequency of Treg cells and an elevated ratio of Treg cells to effector T cells, such as CD8+ T cells, within the TME is consistently associated with an unfavorable prognosis among patients with various cancer types (64, 65). Eliminating Tregs from the tumor environment can thus potentiate the anti-tumor immune response. Moreover, a lower CD8+T/Treg ratio has been identified as a poor prognostic indicator for the effectiveness of anti-PD-1 monoclonal antibody treatments (66). Post-immunotherapy scenarios where there is no appreciable increase in T effector cells coupled with a decrease in Tregs, or a surge in Treg cells within the tumor matrix, are often indicative of resistance to PD-1/PD-L1 monoclonal antibody therapies.
MDSCs, a diverse group of cells, are known to inhibit effector T-cell responses and foster the development of Tregs (67). The efficacy of immunotherapy is often reduced in the presence of the tumor microenvironment (68). These MDSCs are induced in immature myeloid cells by external agents such as tumor-derived factors, and they disrupt the production, proliferation, migration, and activation of MDSCs. MDSCs facilitate tumor invasion and metastasis, predominantly through factors like Indoleamine 2,3-dioxygenase (IDO), Arginase-1 (ARG1), Reactive Oxygen Species (ROS), IL-10, Inducible Nitric Oxide Synthase (iNOS), Cyclooxygenase-2 (COX-2), and Nitric Oxide (NO) (69). Additionally, MDSCs can attract Tregs to the tumor microenvironment to augment immunosuppression. Studies also reveal that inhibiting PI3K can synergize with immunocheckpoint inhibitors. In models where PD-1 monoclonal antibody treatment was ineffective, PI3K inhibition reduced MDSC circulation and recruitment, curtailed the production of immunosuppressive factors like IL-10 and TGF-β, and enhanced the secretion of inflammatory mediators such as Interleukin-12 (IL-12) and Interferon-Gamma (INF-γ), mirroring the combined inhibitory effects on CTLA-4 and PD-1 monoclonal antibodies (70, 71). These findings suggest that PI3K inhibitors could serve as potential adjunctive therapies with PD-1/PD-L1 antibodies, particularly in overcoming single-agent drug resistance. In the metabolic context, MDSCs derive energy from arginine metabolism, primarily through ARG1. Impairment of ARG1 activity can diminish the inhibitory capacity of MDSCs, thereby heightening the sensitivity of tumors to PD-1/PD-L1 antibodies (72).
TAMs, another influential cell type in immunotherapy, consist of M1-like macrophages that bolster anti-tumor immunity and M2-like macrophages that promote cancer. PD-1 expression is more pronounced in M2-like macrophages compared to M1-like macrophages (73, 74), and an increase in PD-1+M2-like macrophages correlates with advanced disease stages, hinting at their progressive accumulation in the tumor microenvironment (75). M2-like macrophages aid in tumor cell immune evasion through PD-1 and are activated by cytokines such as IL-4, IL-10, IL-13, or Colony Stimulating Factor 1 (CSF1), engaging in wound healing, tissue repair, and anti-inflammatory responses through cytokines including IL-10 (76). They also promote tumor invasion and metastasis via angiogenesis and remodeling of the extracellular matrix (77). Clinical studies have correlated high levels of TAMs with poor outcomes in various cancers (78). Targeting the C-C Motif Chemokine Ligand 2 (CCL2) and C-C Motif Chemokine Receptor 2 (CCR2) pathways in a lung adenocarcinoma mouse model led to reduced recruitment of M2 macrophages and inhibited tumor growth (79). Notably, using macrophage Colony Stimulating Factor 1 Receptor (CSF-1R) blockers reduces TAM frequency, increases IFN production, and enhances tumor cell response to drugs in pancreatic cancer models. When combined with PD-1 or CTLA-4 antibodies, and gemcitabine, CSF-1R blockers demonstrated increased efficacy (80).
In conclusion, the heterogeneous nature of inhibitory immune cells within tumors, influenced by chemokines, cytokines, and colony-stimulating factors in the tumor microenvironment, limits the effectiveness of PD-1/PD-L1 blockers when used alone. Resistance to immune checkpoint blockade may be indicated by factors such as the CD8+/Treg ratio, IDO, ARG1, CSF-1R, and the M1/M2 ratio. Addressing these indicators through combined therapeutic strategies could lead to more effective clinical outcomes and prognoses. The concurrent use of drugs targeting immunosuppressive cells, including IDO inhibitors, ARG1 inhibitors, PI3K inhibitors, and ICIs, has shown promise in clinical trials, particularly when used in dual combinations, offering manageable side effects and good clinical compliance. However, the effectiveness of combinations involving three or more such agents remains less explored.
3.3.3 Vascular abnormalities and hypoxia
CD8+ T cells must enter the tumor core through the intratumoral vasculature (16). Their transport into tumor tissue depends on enhanced expression of adhesion molecules and chemokines in the tumor blood vessels, a process known as endothelial cell activation. However, poor activation of tumor blood vessels often leads to impaired transport of CD8+ T cells (81). Studies have shown that the absence of TILs is associated with overexpression of the endothelin B receptor (ETBR) (82). Tumor cells often promote angiogenesis by producing vascular endothelial growth factor (VEGF), which typically reduces the expression of vascular cell adhesion protein 1 (VCAM-1), thereby hindering T cell migration into the TME (83). Additionally, research indicates that Fas ligand (FasL, also known as CD95L) is selectively expressed in the vasculature of human and mouse tumors, whereas it is not expressed in normal vasculature. Expression of FasL enables endothelial cells to kill CD8+ T cells, but not Tregs (84). Tumors with poor vascularization, such as PDAC, due to their abnormal vascular structure and function, reduce the transport of immune cells and often exhibit high resistance to ICIs treatment (85).
Aberrant angiogenesis in tumors frequently precipitates conditions such as hypoxia, acidosis, and necrosis, subsequently impeding anti-tumor immune responses (86). Hypoxia, a defining characteristic of cancer, arises from a disparity between oxygen consumption and supply within the tumor milieu. This is attributed to the voracious oxygen consumption by rapidly proliferating tumor cells, coupled with inadequate oxygen delivery due to dysfunctional vasculature (87). The impact of hypoxia on TILs is complex and wide-ranging. Notably, hypoxia can stimulate the expression of CCL28 (88), VEGF (86), CD39 (89, 90), and CD73 (89, 90). These molecules are instrumental in angiogenesis and modulate T cell mobilization.
3.3.4 Oncogenic pathway activation
In the field of oncology, the complex interplay between tumor cells and various signaling pathways is pivotal in shaping the tumor microenvironment (TME) and influencing therapy resistance. Tumor cells are known to hijack and modulate numerous pathways, notably including PKC, Notch, and TGF-β signaling. Recently, attention has also been drawn to the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) and Siglec signaling pathways. These pathways play a critical role in sustaining a tumor-friendly microenvironment and fostering resistance to treatment, including multi-drug resistance.
3.3.4.1 Protein kinase C signaling
In oncology, the role of PKC isoforms in the TME is increasingly recognized as critical in determining tumor behavior. PKC, a family of serine/threonine kinases, serves as a signal transducer for various molecules including hormones, growth factors, cytokines, and neurotransmitters. These molecules are key regulators of cell survival, proliferation, differentiation, apoptosis, adhesion, and malignant transformation (91–93). The interaction of ligands with receptors can activate phospholipase C, thereby upregulating activators of PKC signaling like diacylglycerol (DAG) and Ca++ (94, 95), subsequently modulating several molecular pathways such as Akt, STAT3, NF-κB, and apoptotic pathways. Interestingly, different PKC isoforms play varying roles in tumorigenesis and metastasis (94). For instance, PKC alpha demonstrates antitumor activity by influencing the polarization of TAMs within the TME (96). Conversely, PKC theta has been shown to suppress tumors by inducing immune suppression through CTLA4-mediated regulatory T-cell function (97–99). However, other isoforms, like PKC beta, are known to facilitate angiogenesis and invasiveness in certain tumors via the VEGF signaling pathway (100–102). The complexity of PKC signaling is further evidenced by its dual role as both a tumor promoter and suppressor, depending on the isoform and the context. This dual role presents both challenges and opportunities for therapeutic interventions.
Recent advancements in cancer treatment strategies have explored the modulation of PKC signaling, utilizing activators like Bryostatins (103) and Epoxytiglianes (104–106), as well as inhibitors such as CGP 41251, to counteract tumor growth and reverse multidrug resistance (107).
3.3.4.2 PI3K-AKT-mTOR signaling pathway
The PI3K/AKT/mTOR signaling pathway, a pivotal regulator of cellular processes such as apoptosis, proliferation, movement, metabolism, and cytokine expression, plays a critical role in tumor development and resistance mechanisms, especially against PD-1/PD-L1 antibodies. Central to this pathway is the lipid phosphatase PTEN, a tumor suppressor that inhibits PI3K activity. PTEN deletion or mutation leads to the activation of PI3K/AKT and resistance to PD-1/PD-L1 in various cancers. PTEN’s expression is regulated through diverse mechanisms, including epigenetic silencing, post-transcriptional and post-translational modifications, and protein-protein interactions (108). PTEN’s downregulation is key in cancer progression, affecting cell energy metabolism, metabolic reprogramming of cancer cells, and influencing glucose uptake and protein synthesis. PTEN also plays a role in cell migration and senescence, with its loss leading to increased cell viability and promoting EMT and tumor cell migration (109). PTEN loss affects tumor immunotherapy, showing a correlation with resistance to immunotherapy, particularly impacting the tumor microenvironment (110). PTEN deficiency leads to downregulation of SHP-2, a negative regulator of JAK/STAT3 pathway, promoting tumor growth (111, 112). The loss of PTEN in certain cancers is associated with decreased T-cell function, increased VEGF production, and the release of anti-inflammatory cytokines, resulting in non-inflammatory tumors. Moreover, PTEN’s role extends to regulating PD-L1 levels, with its absence or constitutive expression of the PI3K/AKT pathway influencing PD-L1 expression (113, 114). This interaction affects PD-1/PD-L1 antibody responses and is subject to modulation by various intracellular signaling pathways, including RAS/RAF/MEK and JAK/STAT, influenced by IFN-γ released by immune cells. Selective inhibition of PI3K has shown to enhance the therapeutic effect of PD-1/PD-L1 and CTLA-4 antibodies in experimental models, indicating potential in reversing resistance to immunocheckpoint inhibitors (115). Further clinical studies are warranted to explore this possibility.
3.3.4.3 TGF-β signaling
Transforming Growth Factor-Beta (TGF-β) plays a multifaceted role in the progression of cancer, affecting a variety of cellular processes including cell proliferation, angiogenesis, epithelial-to-mesenchymal transition, immune infiltration, metastatic dissemination, and drug resistance (116). Interestingly, TGF-β produced by tumor cells can alter the function of tumor-associated plasmacytoid dendritic cells (pDCs), particularly affecting their ability to produce Type I interferon, thereby impacting T cell recruitment (117, 118). This aspect of TGF-β signaling is crucial in understanding its role in excluding T cells from the TME.
Recently, the combined use of TGF-β blocking antibodies with PD-L1 antibodies has been proven effective in enhancing T cell penetration into tumors, boosting anti-tumor immunity, and leading to tumor regression (44). Additionally, TGF-β signaling plays a dual role in cancer progression. Initially, it acts as a tumor suppressor by inhibiting cell proliferation and inducing apoptosis (119). However, as malignancies progress, cancer cells exploit TGF-β signaling to create a favorable TME, activating CAFs, promoting angiogenesis, and suppressing anti-tumor immune responses (120–122). Given its complex role, the side effects of targeting TGF-β signaling in therapeutic interventions are also a concern.
3.3.4.4 cGAS-STING signaling
Recent advancements in cancer research have increasingly focused on the cGAS-STING signaling pathway and its role in tumor progression. Analysis of The Cancer Genome Atlas (TCGA) database, which classifies 18 different types of malignant tumors, has revealed variations in the expression of key genes involved in the cGAS-STING signaling mechanism between normal and cancerous tissues. These include genes encoding cGAS (MB21D1), STING (TMEM-173), TBK-1, and IRF-3. Studies have found that these genes are significantly upregulated in nearly all cancer models, indicating a possible universal activation of cGAS-STING signaling in various cancer types (123, 124). Interestingly, some highly invasive tumors seem to rely on the cGAS-STING pathway to facilitate tumorigenesis, impacting cancer treatment approaches (125, 126). The NF-κB pathway, known for regulating cell proliferation, apoptosis, and survival, also plays a vital role in the inflammatory response. Its activation can contribute to inflammation, tumor development, and immune dysfunction. Chromosomal instability can lead to chronic inflammation by persistently activating the cGAS-STING pathway, which in turn enhances NF-κB function and promotes the progression of metastatic cancer cells (126, 127).
Furthermore, TCGA data analysis has demonstrated a negative correlation between STING expression levels in cancer and the infiltration of immune cells in various tumor models. This suggests that an increase in cGAS-STING signaling may predict poorer outcomes in cancer patients (123). Additionally, certain tumor cells promote brain metastasis by enhancing astrocyte-gap junctions through the expression of PCDH7 (composed of Cx43). These junctions transfer cGAMP from cancer cells to adjacent astrocytes, activating STING and triggering the production of TNF and IFN-α. These paracrine signals further activate NF-κB and STAT-1 pathways in metastatic brain cells, contributing to brain metastasis and resistance to lung and breast cancer therapies (128).
4 Therapeutic strategies for cold tumors
4.1 Dual ICIs
4.1.1 α−CTLA−4 combined with α−PD−1/PD−L1
T cell activation requires two essential signals: the T cell receptor (TCR) and costimulatory pathways (129). Numerous costimulatory receptors have been discovered, which bidirectionally regulate T cell responses (130). Identified as the first molecule to deliver inhibitory signals, CTLA-4 is critical for concluding immune responses (131, 132). It negatively regulates T cell activation by, for instance, competing with CD28 for binding shared ligands B7.1 and B7.2 (133).
In clinical therapeutics, ipilimumab is seldom used in isolation. Physicians typically administer it in combination with nivolumab. While both CTLA-4 and PD-1 serve as immune checkpoints that inhibit T-cell activation via distinct mechanisms, their modulatory effects on immune response are uniquely characterized. Consequently, anti-CTLA-4 monoclonal antibodies may synergize with anti-PD-1/PD-L1 counterparts to potentiate tumor immunity. A growing body of research indicates that dual blockade of PD-1/PD-L1 and CTLA-4 exhibits enhanced antitumor efficacy in certain cancer types (134). Studies from CheckMate-069, CheckMate-067, and CheckMate-142 demonstrate that the combination of ipilimumab and nivolumab significantly improves clinical outcomes compared to monotherapy with either agent alone (135–137). Data from CheckMate-214, CheckMate-227, and CheckMate-743 further reveal superior treatment efficacy of the ipilimumab plus nivolumab regimen over standard targeted or chemotherapy approaches (138–140). To date, the U.S. FDA has approved the use of the ipilimumab and nivolumab combination for the treatment of melanoma, renal cell carcinoma, microsatellite instability-high/mismatch repair-deficient colorectal cancer, hepatocellular carcinoma, PD-L1 positive non-small cell lung cancer, and malignant pleural mesothelioma (134–140).
4.1.2 α−PD−1/PD−L1 combined with ICIs
Emerging dual immune checkpoint blockade strategies, encompassing the combination of α-PD-1/PD-L1 with α-TIM-3, α-LAG-3, α-PVRIG, and α-TIGIT, remain in clinical trials and await regulatory approval. The ligation of TIM-3 to galectin-9 instigates apoptosis in Th1 cells via calcium flux (141). This dual inhibition, when applied to TIM-3 and PD-1/PD-L1 pathways, markedly augments anti-tumor immunity, as shown by slower tumor growth in murine models (142). Data from clinical trials suggest that this combined blockade does not increase adverse effects, although optimization of patient selection is warranted (143–145).
Extending the scope of ICIs, α-LAG-3, α-PVRIG, α-TIGIT, and α-Siglec-10, when used in concert with α-PD-1/PD-L1, enhance TIL functionality and concomitantly inhibit tumor growth (146–149). The RELATIVITY-047 phase 2/3 trial revealed a notable PFS advantage with relatlimab (α-LAG-3) plus nivolumab in late-stage melanoma (10.1 vs. 4.6 months; HR: 0.75), outperforming nivolumab alone (150). Additionally, COM701 (α-PVRIG) with nivolumab exhibited promising antineoplastic activity in phase 1 trial NCT03667716, inclusive of patients with prior ICI treatment (151). In phase 2 trial NCT03563716, tiragolumab (α-TIGIT) plus atezolizumab significantly improved both response rates (OR: 2.57, 95%CI: 1.07–6.14) and PFS (HR: 0.57; 95%CI 0.37–0.90) in PD-L1 positive NSCLC patients, compared with the control group receiving placebo and atezolizumab (152).
4.2 CAR-T cell therapy combined with ICIs
Chimeric Antigen Receptors (CARs) are multifaceted constructs, typically encompassing an extracellular antigen-binding domain, such as a single-chain variable fragment (scFv) targeting CD19, a hinge region to enhance antigen-receptor and tumor antigen interaction, a transmembrane domain for functional stability, and a T-cell activation domain (CD3 ζ) for primary signaling. Additionally, one or more intracellular co-stimulatory domains, like CD28/4-1BB, are included for secondary T-cell activation signaling (153). CAR T-cell activation is contingent on the presence of TAAs or TSAs. CARs’ unique ability to recognize diverse targets, including both protein and non-protein entities, on the cell surface, activates T cells without the necessity for antigen processing and presentation. This capability, bypassing human MHC constraints, positions CAR T-cell therapy as a revolutionary approach in T-cell therapeutic strategies, noted for its distinctive treatment characteristics (154).
CAR-T cell therapy’s hallmark is its non-reliance on Major Histocompatibility Complex (MHC) restrictions, coupled with an enhanced tumor-specific immune response, facilitated by the incorporation of co-stimulatory domains such as CD28, OX40, and 4-1BB. This attribute offers the potential to effectively target ‘cold tumors’, characterized by limited pre-existing T cell infiltration and a paucity of tumor antigens. A multitude of ongoing clinical trials are exploring CAR T-cell therapies against solid tumors, as elaborated in Table 1. Despite the direct tumor cell eradication capabilities of CAR-T cells, they remain susceptible to immunosuppression via immune checkpoints. Consequently, the synergistic approach of integrating ICIs with CAR-T cell therapy emerges as a promising treatment strategy (155, 156).
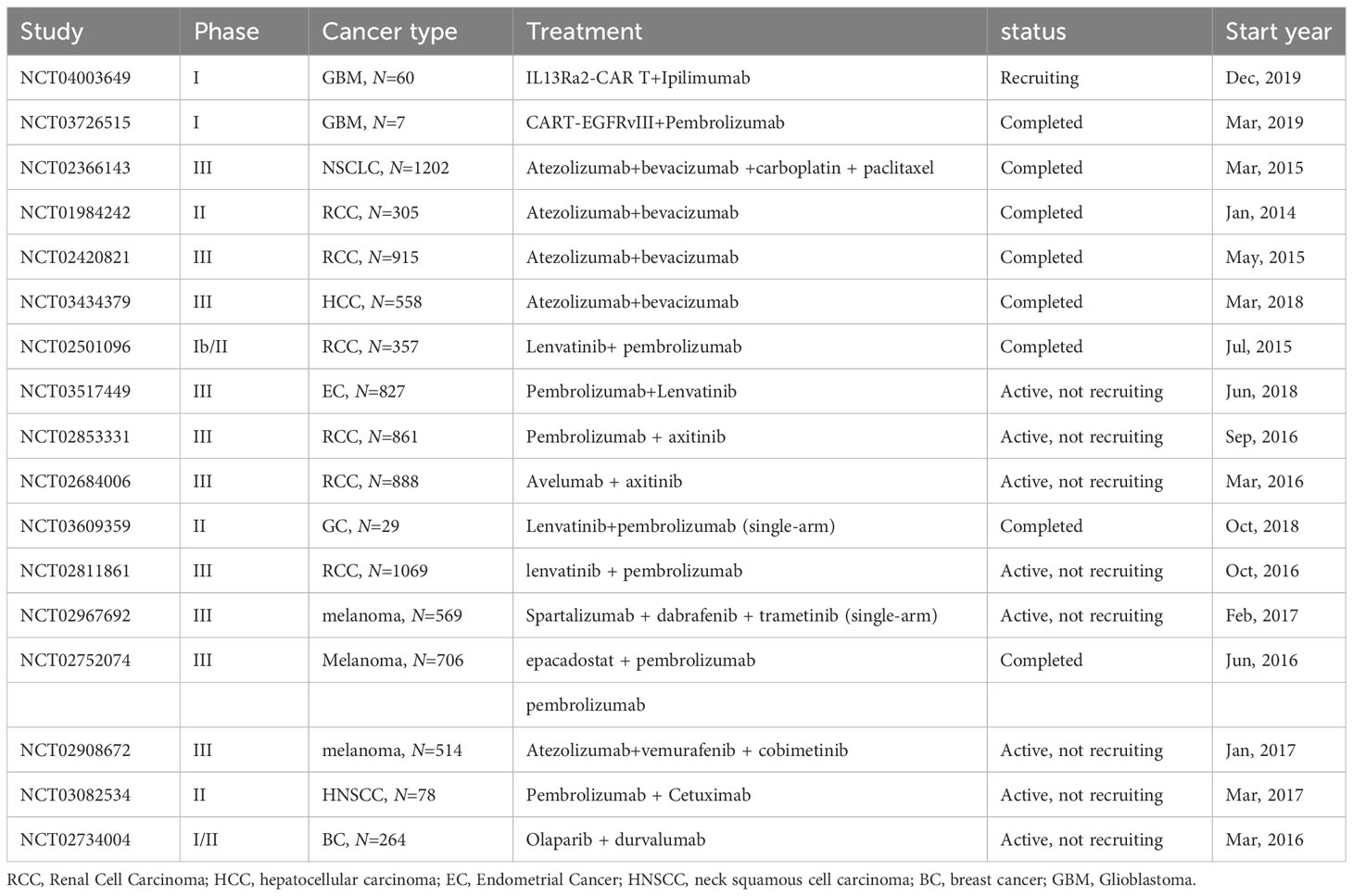
Table 1 Key clinical trials of immunotherapy combined with CAR-T cell/targeted therapy.
4.3 CAR-NK cell therapy
A noteworthy attribute of mature NK cells in the field of adoptive cell therapy is their ability to retain functionality when transplanted into new environments with differing MHC expression patterns (157, 158). Unlike T lymphocytes, NK cells predominantly do not trigger graft-versus-host disease but instead exert a regulatory role (159). Advances in genetic modification techniques have shown that NK cells can be customized further, including the introduction of CARs and the knockout of inhibitory genes (160). These advancements enable NK cells from patients with hematologic malignancies to rapidly eliminate autologous tumor cells resistant to unmodified NK cells (161, 162). Preclinical studies on CAR-NK cells in xenograft mouse models have demonstrated in vivo activity comparable to CAR-T cells, yet with less cytokine release and improved overall survival rates (163, 164). The inaugural human study of CAR-NK cells has revealed promising anti-tumor responses without significant toxicities such as cytokine release syndrome and graft-versus-host disease (165). These positive outcomes lay the groundwork for further development of CAR-NK cells as a promising modality for cancer therapy (162).
Currently, CAR-NK cell-mediated immunotherapy is advancing rapidly, offering new therapeutic avenues for patients with malignant tumors. Despite extensive research in the field of cancer immunotherapy, the application of CAR-NK cells remains relatively limited to a variety of tumor models, primarily focusing on hematological malignancies (166). Table 2 summarizes the clinical studies of CAR-NK cells in solid tumors.
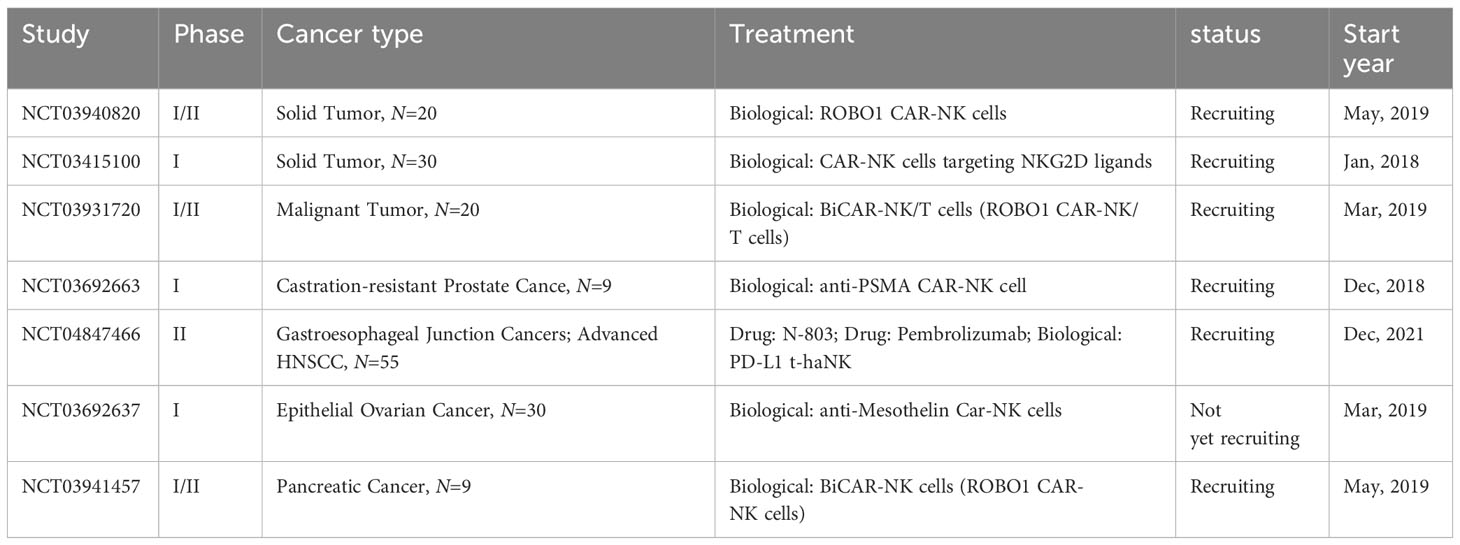
Table 2 Clinical trials of CAR-NK cell-based cancer immunotherapy.
4.4 T Cell redirecting bispecific antibody combined with ICIs
T cell-redirecting bispecific antibodies (BsAbs) represent a cutting-edge approach in immunotherapy, merging two monoclonal antibodies into a singular entity. Ingeniously engineered, these antibodies concurrently engage specific receptors on T cells, like CD3, and distinct tumor cell antigens. Central to their dual-specific functionality is the ability to directly steer T cells towards tumor cells, thus enhancing T cell-mediated identification and elimination of tumor cells. A distinctive feature of BsAbs-induced tumor cell lysis is its independence from conventional antigen recognition processes, which typically involve MHC class I or II molecules, antigen-presenting cells, or the necessity of co-stimulatory molecules (167).
Advancements in T cell-redirecting BsAbs for solid tumors lag behind those in hematological malignancies, in part due to a more limited range of available surface targets in solid tumors (167). Despite these challenges, four bispecific antibodies (BsAbs) have currently received FDA approval. These include Catumaxomab (Fresenius/Trion’s Removab®), which was withdrawn from the market in 2017, Blinatumomab (Amgen’s Blincyto®), Amivantamab-vmjw (Janssen’s Rybrevant®), and Tebentafusp-tebn (Immunocore’s Kimmtrak®) (168). In addition, there are still many BsAbs in the clinical evaluation stage for cancer treatment (Table 3). These agents signify progress in targeted therapeutic interventions, illustrating the evolving landscape of cancer treatment. However, even with their effectiveness, the T cells activated by these therapies can be rendered inactive by immune checkpoints. Consequently, a synergistic approach of ICIs in conjunction with T cell-redirecting BsAbs presents as a viable and potentially more effective treatment strategy.
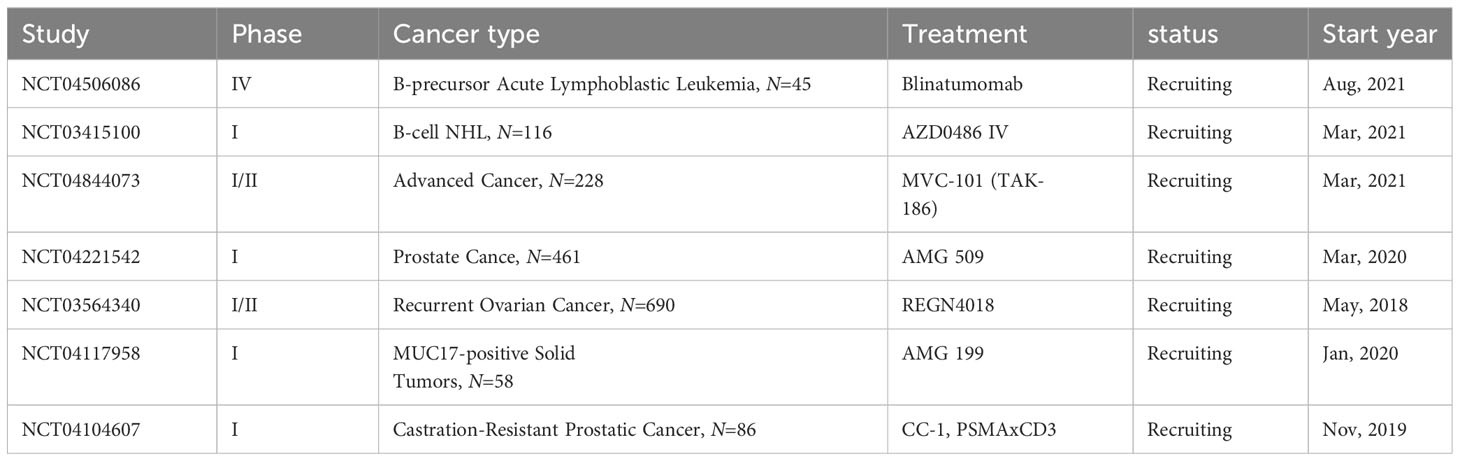
Table 3 Bispecific antibody clinical trials ongoing.
4.5 Cancer vaccine
‘Cold’ tumors, characterized by a dearth of tumor antigens, commonly exhibit immune evasion. Nonetheless, the use of cancer vaccines containing tumor antigens has shown efficacy in eliciting immune responses against such tumors (169). A range of cancer vaccines, designed to bolster the patient’s immune system, have received approval, including Tedopi, Ilixadencel, GVAX, and PolyPEPI101884 (170). Notably, Sipuleucel-T is the first FDA-approved cancer vaccine for metastatic castration-resistant prostate cancer (mCRPC), significantly prolonging patient survival (171). However, the therapeutic efficacy of cancer vaccines is often impeded by high PD-1 expression in effector T cells (172, 173). To address this, numerous phase 1 clinical trials have been initiated to investigate the combined use of cancer vaccines and immunoglobulins in cancer treatment, demonstrating their combined potential (174, 175). Ongoing clinical trials in this domain include NCT04300244, NCT03632941, KEYNOTE-603, and NCT03743298.
4.6 Oncolytic virus combined with ICIs therapy
Oncolytic viruses, encompassing both natural and genetically engineered variants, induce tumor cell lysis by selectively infecting and proliferating within tumor cells. Beyond their direct antitumor activity, these viruses also provoke a comprehensive, potent, and enduring anti-tumor immune response. This response is facilitated by the liberation of TAAs and additional DAMPs upon tumor cell demise (176). A significant aspect of oncolytic viral therapy is its systemic immunomodulatory impact, which extends its effects beyond the injection locus to distant tumor regions (176).
T-VEC, a modified herpes simplex virus, demonstrates augmented anti-tumor efficacy in treating unresectable stage IIIB-IV melanoma when used in conjunction with Ipilimumab, surpassing the results achieved with Ipilimumab alone (177). Furthermore, integrating a PD-1 inhibitor with oncolytic viral therapy significantly boosts its anti-tumor potency in glioma models (178). In the context of triple-negative breast cancer (TNBC), the response to ICIs typically remains suboptimal. Oncolytic viral treatment, however, renders TNBC more responsive to immune checkpoint blockade, successfully averting recurrence in a majority of the treated animal models (179).
4.7 Macrophage targeted therapy combined with ICIs therapy
TAMs, as key immune constituents in the TME, play an integral role in solid tumor development. These cells exhibit dual phenotypes: anti-tumoral (M1) and pro-tumoral (M2), with their behavior governed by their polarization state (180). TAMs significantly modulate immune responses by producing an array of cytokines and effector molecules. They suppress the function of T cells, B cells, NK cells, and dendritic cells, while simultaneously enhancing the roles of Tregs, T helper 17 cells (Th17), γδ T cells, and MDSCs. This multifaceted approach fosters an immunosuppressive milieu within the TME (181). Crucially, TAMs’ association with PD-L1 expression suggests that strategies combining ICIs with targeted TAM therapies could offer substantial therapeutic benefits (182, 183).
TAMs are pivotal in cancer treatment strategies. Targeting TAMs typically involves three approaches: 1) eradicating existing TAMs in the TME, 2) curtailing the recruitment of monocytes, and 3) reprogramming TAMs (181). TAMs are pivotal in cancer treatment strategies. Targeting TAMs typically involves three approaches: 1) eradicating existing TAMs in the TME, 2) curtailing the recruitment of monocytes, and 3) reprogramming TAMs (184, 185). In a phase 1b study (NCT02323191), a combination of the CSF-1R inhibitor emactuzumab with atezolizumab exhibited a superior Objective Response Rate (ORR) compared to controls (186). Additionally, in a separate clinical trial, the C-C chemokine receptor type 5 (CCR5) inhibitor maraviroc, used in tandem with pembrolizumab, demonstrated notable efficacy in patients with dMMR CRC (187).
4.8 Radiotherapy combined with ICIs therapy
Radiation therapy, employing ionizing radiation to directly destroy tumor cells, exerts multifaceted impacts on tumor immunity: 1) It can trigger immunogenic cell death (ICD) in tumor cells, culminating in the release of abundant DAMPs. These DAMPs, once phagocytosed by DCs, facilitate DC maturation (188). 2) Mature dendritic cells are capable of cross-presenting tumor antigens to CD8+ T cells, thereby initiating specific immune responses (189). 3) Concurrently, radiation therapy exhibits immunosuppressive properties, encompassing bone marrow suppression, the direct eradication of immune cells, upregulation of immune checkpoints, and the elicitation of immunogenic cytokines and chemokines (190–192). These immunoregulatory effects lay the groundwork for integrating ICIs with radiation therapy.
In certain cases, patients undergoing combined therapies exhibit spontaneous tumor regression beyond the irradiated zones, termed the ‘abscopal effect’ or radiotherapy’s distant impact. This phenomenon is largely attributed to radiotherapy enhancing the antigen presentation of tumor cells, thereby augmenting CD8+ T cell production. These cells then travel via the bloodstream to remote sites, influencing tumors outside the irradiated areas (193, 194). Contemporary research suggests that the abscopal effect can counteract immunosuppression and enhance the efficacy of ICIs (195–198). For instance, preliminary results from a phase III study on stage III unresectable NSCLC patients revealed that post-radiotherapy treatment with durvalumab significantly prolonged progression-free survival (PFS) compared to a placebo (199). Furthermore, another phase III study involving 799 participants demonstrated that, in patients with mCRPC previously treated with docetaxel, a combination of ipilimumab and radiotherapy markedly increased overall survival (OS) over placebo plus radiotherapy (200). Additional information on clinical trials combining radiotherapy with ICIs is detailed in Table 4.
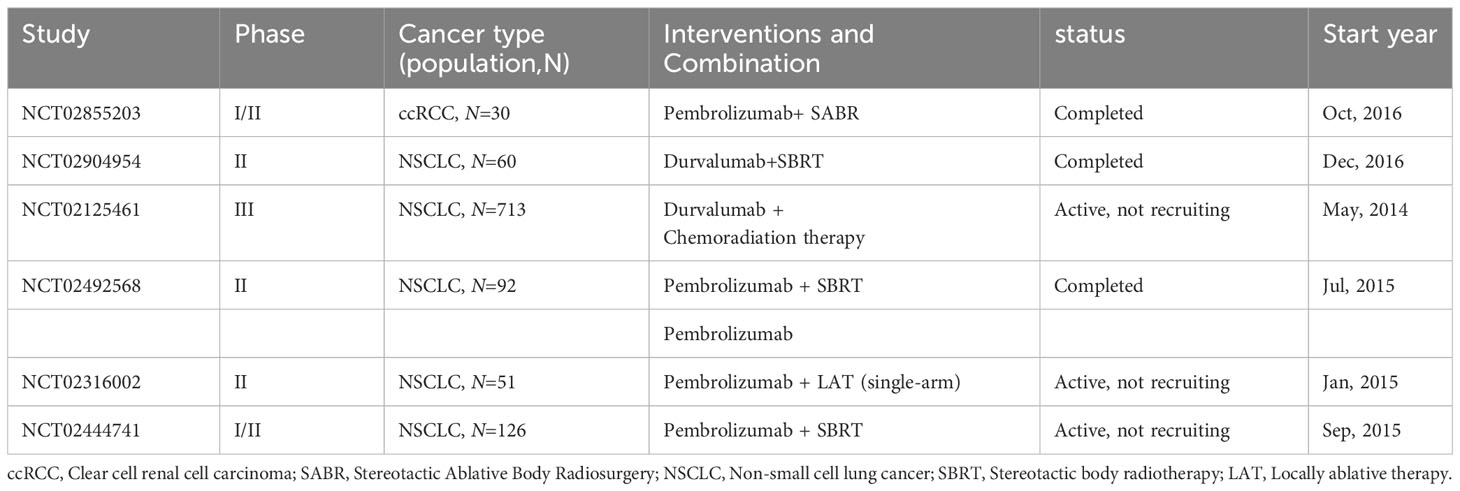
Table 4 Key clinical trials of immunotherapy combined with radiotherapy.
4.9 Chemotherapy combined with ICIs therapy
Chemotherapy drugs wield a bidirectional impact on the immune system during tumor therapy. Initially, they frequently induce systemic immunosuppression, evident through bone marrow suppression and lymphocyte depletion. Concurrently, these agents can eradicate specific immune cells, contributing to the reconstitution and establishment of a renewed immune system (201). The immunostimulatory actions of chemotherapy are manifested in several ways: 1) Augmenting antigenicity: Agents like cyclophosphamide, gemcitabine, platinum-based drugs, and taxanes boost the antigenicity of tumor cells. 2) Increasing susceptibility to immune assaults: This is primarily achieved by improving the visibility of tumor cells to the immune system. 3) Triggering ICD and antigen-specific responses: Anthracyclines, mitoxantrone, and oxaliplatin accomplish this by interacting with DNA replication and repair mechanisms (202). These pathways illustrate that chemotherapeutic drugs not only directly eradicate tumor cells but also engage in combatting tumors by stimulating and modulating the immune system.
Clinical trials integrating chemotherapy with immunotherapy have demonstrated considerable therapeutic success. In the phase III KEYNOTE-189 trial (NCT02578680), a regimen of pembrolizumab combined with pemetrexed-platinum agents yielded an ORR of 48.3%, markedly surpassing the 19.9% ORR of the placebo plus pemetrexed-platinum cohort. This combination therapy also significantly enhanced OS with a Hazard Ratio (HR) of 0.56 (95% Confidence Interval [CI]: 0.46-0.69) and PFS with an HR of 0.49 (95% CI: 0.41-0.59) (203). The phase III KEYNOTE-355 trial revealed that supplementing standard chemotherapy with pembrolizumab substantially improved PFS in metastatic TNBC patients with a combined positive score (CPS) of 10 or above (204). The KEYNOTE-021 (NCT02039674) trial found that pembrolizumab plus chemotherapy exhibited superior ORR (58% vs 33%) and PFS (median of 24.5 months vs 9.9 months; HR: 0.54; 95% CI: 0.35-0.83) compared to chemotherapy alone (205). Furthermore, in the phase III IMpower133 trial (NCT02763579), the addition of atezolizumab to carboplatin and etoposide significantly prolonged OS and PFS in small-cell lung cancer patients over the placebo with carboplatin and etoposide (206). Other clinical trials of chemotherapy combined with ICIs treatment are detailed in Table 5.
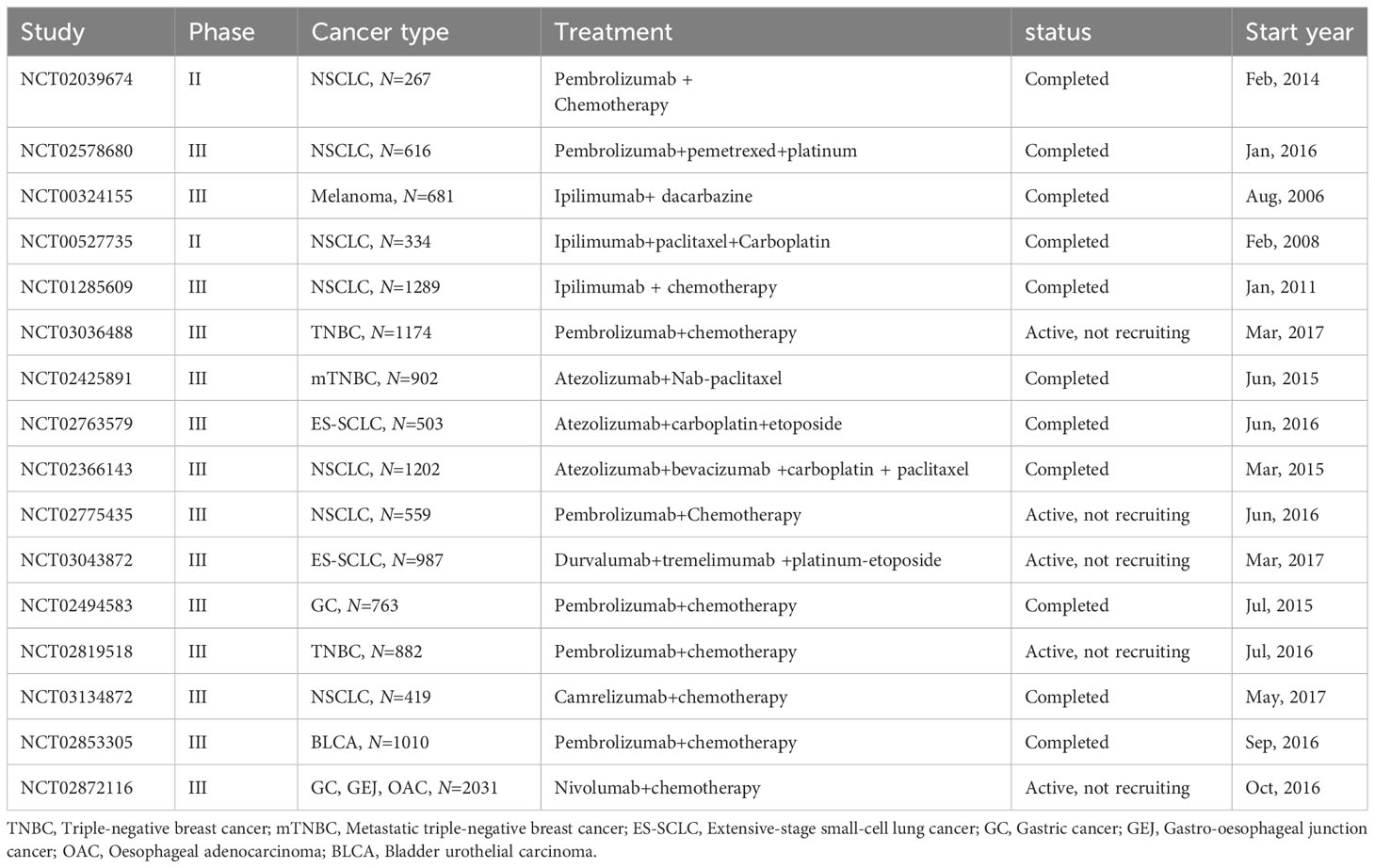
Table 5 Key clinical trials of immunotherapy combined with chemotherapy.
4.10 Targeted therapy combined with ICIs therapy
Targeted cancer therapy is predicated on creating potent inhibitors that specifically target molecular markers of tumor cells, aiming to effectively treat the cancer. The modes of action of this therapy span a range, including suppressing tumor cell proliferation, intervening in the cell cycle, promoting tumor cell differentiation, curbing metastasis, inducing apoptosis, and hampering tumor angiogenesis (207). Despite the considerable successes of many targeted therapies in clinical settings, the emergence of drug resistance in a significant number of patients represents a formidable challenge. Recent research has revealed that these targeted agents can trigger ICD in tumor cells, thereby bolstering the effectiveness of ICIs (207). Consequently, integrating targeted therapy with immunotherapy emerges as a novel and promising approach to surmount drug resistance and enhance therapeutic outcomes.
In the phase III IMspire150 trial (NCT02908672), the combination of vemurafenib, cobimetinib, and atezolizumab was compared against a control regimen of vemurafenib, cobimetinib, and placebo in patients with advanced or metastatic melanoma harboring the BRAF V600 mutation. The study revealed that the addition of atezolizumab significantly extended PFS to 15.1 months, compared to 10.6 months in the control group, with a HR of 0.78 (95% CI: 0.63-0.97, p=0.025) (208). In the phase 1/2 MEDIOLA trial, the efficacy of Olaparib combined with durvalumab was evaluated in patients with metastatic breast cancer with germline BRCA1 or BRCA2 mutations. This trial showed positive safety and disease control outcomes in 80% of patients after 12 weeks of treatment. These findings underscore the potential of integrating targeted therapy with immunotherapy in the treatment of certain cancers (209). Detailed information on additional clinical trials combining targeted therapy with ICIs is available in Table 1.
4.11 STING agonist combined with ICIs therapy
The accumulation of cytosolic chromatin fragments and micronuclei, a hallmark of malignant transformation in cancer cells, raises the likelihood of cytosolic DNA escape or DCs engulfing tumor-derived DNA (210). The cGAS-STING pathway, pivotal for cytosolic DNA detection, plays a crucial role in this context. Binding of cytosolic double-stranded DNA (dsDNA) to cGAS triggers the synthesis of cyclic GMP-AMP (cGAMP). This activation leads to the transformation of STING from a monomer to a dimer, facilitating its relocation from the endoplasmic reticulum to perinuclear microsomes. Subsequently, STING engages and phosphorylates TBK1, initiating a cascade that activates IRF3 and boosts IFN-I production (211–213). Additionally, the STING pathway enhances IFN-I through the NF-κB route (214). IFN-I, a multifaceted immune enhancer, significantly augments the functions of DCs, NK cells, and T cells (215). The cGAS-STING pathway’s integral role in linking innate and adaptive immunity underscores its potential as a target for cancer immunotherapy.
Initial clinical trials with Dimethyloxoxanthenyl acetic acid (DMXAA), the first STING agonist, were unsuccessful (216). Further research revealed that DMXAA specifically activates the mouse STING pathway, with minimal effects on its human counterpart (217, 218). Consequently, several natural and synthetic cyclic dinucleotides (CDNs), structurally and functionally akin to cGAMP, have emerged as promising STING agonists in cancer immunotherapy (219–221). However, these CDNs typically face challenges like limited transmembrane transport and reliance on intratumoral injection. Recent developments include novel STING agonists like diABZI and MSA-2, which offer systemic administration possibilities (222, 223). Moreover, manganese has been identified as a natural STING agonist, playing a significant role in anti-tumor immunity (224, 225).
In the context of combination therapy, the synergy of STING agonists with α-PD-1/PD-L1 antibodies presents a promising avenue. This approach simultaneously amplifies innate and adaptive immunity, effectively countering immunotherapy resistance. STING agonists enhance immune cell infiltration and amplify the functionality of APCs, NK cells, and T cells (226–228). Concurrently, α-PD-1/PD-L1 antibodies capitalize on the PD-L1 upregulation induced by STING agonists (227). Ongoing clinical trials involving combinations like ADU-S100 with spartalizumab, MK-1454 with pembrolizumab, and manganese with α-PD-1 have shown promising anti-tumor efficacy and tolerable safety profiles (229, 230).
4.12 Nanoparticles combined with ICIs therapy
Rapidly proliferating cancer tissues frequently form enlarged vascular endothelial gaps, with an average size of several hundred nanometers, to draw more nutrients from the body. Such gaps are generally not found in normal tissues. This phenomenon, known as the enhanced permeability and retention effect (EPR), allows nanoparticles of suitable size to infiltrate tumor tissues, while being restricted by the denser structure of normal tissues. This underpins the theoretical foundation for targeting tumor tissues with nanoparticles (231). Nanoparticles can amplify the efficacy of immunotherapy by inducing ICD in tumor cells (232). In melanoma mouse models, pH-sensitive liposomes equipped with a dual delivery system of doxorubicin hydrochloride and deferasirox have shown to enhance antigen presentation and T-cell infiltration, thereby augmenting their anti-tumor action (233). In glioblastoma multiforme (GBM) research, where drug delivery is constrained by the blood-brain barrier, BAMPA-O16B/siRNA liposomes have been able to effectively transport anti-CD47 and PD-L1 siRNA into intracranial GBM tumors in mice (234). Consequently, the strategy of using nanoparticles in combination with ICIs represents a promising avenue of research.
5 Conclusion and perspectives
In the evolving landscape of oncology, ICIs have emerged as a pivotal advancement. However, their efficacy is not uniform across all patient groups, highlighting a need for more nuanced understanding beyond the simplistic ‘cold’ and ‘hot’ tumor classifications. Intriguingly, some ‘hot’ tumors show responsiveness to ICIs despite a scarcity of CD8+ T cells, driven by NK cell-mediated immune responses. Conversely, ‘cold’ tumors often struggle with T cell activation and infiltration issues. Strategies that combine ICIs with other treatments such as radiotherapy, chemotherapy, CAR-T cell therapy, or targeted therapy are being investigated to transform ‘cold’ tumors into ‘hot’ ones, potentially increasing the efficacy of ICIs. Currently, many combination therapies fail to replicate these results in clinical settings. Currently, only a limited number of combinations, including α-PD-1/PD-L1 with chemotherapy, angiogenesis inhibitors, or α-CTLA-4, have received regulatory approval. The efficacy of most combinations remains confined to animal tumor models, underscoring the need for optimal preclinical models, with humanized patient-derived models offering more precise efficacy evaluations. However, combination therapies pose challenges such as increased immune-related adverse events (irAEs) and healthcare costs, and the risk of exposing patients to higher toxicities with inappropriate combinations. Optimizing administration regimens, including dosage, timing, and sequence, is crucial for the development of these therapies. Furthermore, the selection of suitable combination therapies and identification of predictive biomarkers for treatment response are still areas of active investigation. Liquid biopsy, by monitoring the dynamic immune landscape of the tumor microenvironment, offers a promising approach for real-time biomarker identification, guiding precision immunotherapy. Personalized combination therapies based on immune profiling and other predictive biomarkers, and a comprehensive framework integrating genomic, transcriptomic, immune, and microbiome profiles, could enhance patient selection for combination treatments. Particularly for patients with ‘cold’ tumors, α-PD-1/PD-L1 monotherapy often falls short of clinical benefits, necessitating personalized combinations to overcome drug resistance. In immune-desert scenarios, treatments such as radiotherapy, chemotherapy, and STING agonists can counter low immunogenicity-mediated immune tolerance by inducing immunogenic cell death and promoting antigen-presenting cell function. These combinations with α-PD-1/PD-L1 can simultaneously enhance multiple aspects of the cancer-immunity cycle, reshape the tumor microenvironment, and facilitate the transformation from non-inflamed to inflamed tumors. Additionally, the development of next-generation α-PD-1/PD-L1 drugs, including bifunctional or bispecific antibodies, could extend the indications for α-PD-1/PD-L1 therapies, allowing a broader range of patients to benefit from these advanced treatments.
Author contributions
PO: Visualization, Writing – original draft. LW: Visualization, Writing – original draft. JW: Visualization, Writing – original draft. YT: Writing – original draft. CC: Writing – original draft. DL: Writing – review & editing. ZY: Writing – review & editing. RC: Writing – review & editing. GX: Writing – original draft. JG: Writing – original draft. ZB: Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Fundamental Research Funds for the Central Universities (21623305,21623409), Guangdong Medical Science and Technology Research Fund Project(A2023398) and Guangzhou Science and Technology Plan City-School Joint Funding Project (202201020304).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CAFs, cancer-associated fibroblasts; CCR5,C-C motif chemokine receptor 5; cDC1, type 1 classical DC; cDC2, type 2 classical DC; CI, confidence interval; CPS, combined positive score; CRC, colorectal cancer; CRT, calreticulin; cSCC, cutaneous squamous cell carcinoma; CSF-1R, colony-stimulating factor 1 receptor; CTLs, cytotoxic T-lymphocytes; CXCR3, CXC-chemokine receptor 3; DCs, dendritic cells; dMMR, defective mismatch repair; DNMT1, DNA methyltransferase 1; ECM, extracellular matrix; EPR, enhanced permeability and retention effect; ETBR, endothelin B receptor; EZH2, enhancer of zeste homologue 2; FasL, Fas ligand; GBM, glioblastoma multiforme; HLA-I LOH, HLA-I loss of heterozygosity; HR, hazard ratio; ICD, immunogenic cell death; ICIs, immune checkpoint inhibitors; mCRPC, metastatic castration-resistant prostate cancer; MDSCs, myeloid-derived suppressor cells; mIF, multiplex immunofluorescence; MIS-H, high microsatellite instability; MMRp, mismatch repair proficient; NK, natural killer; NSCLC, unresectable non-small cell lung cancer; NSSMs, nonsynonymous somatic mutations; ORR, objective response rate; OS, overall survival; OV, oncolytic viruses; PD-1, programmed death-1; PDAC, pancreatic ductal carcinoma; pDCs, plasmacytoid DCs; PFS, progressionfree survival; PRC2, polycomb repressive complex 2; RNS, reactive nitrogen species; scRNA-seq, single-cell RNA sequencing; sGSN, secreted gelsolin; TAAs, tumor-associated antigens; TAMs, tumor-associated macrophages; VCAM-1, vascular cell adhesion protein 1; TCR, T cell receptor; VEGF, vascular endothelial growth factor; TGFβ, transforming growth factor β; Th1, type 1 helper T cells; Th2, type 2 helper T cells; TILs, tumor-infiltrating T lymphocytes; TMB, tumor mutational burden; TME, tumor microenvironment; TNBC, triplenegative breast cancer; Treg, regulatory T cells; TSAs, tumor-specific antigens; TVEC, Talimogene laherparepvec.
References
1. Zhang J, Huang D, Saw PE, Song E. Turning cold tumors hot: from molecular mechanisms to clinical applications. Trends Immunol. (2022) 43:523–45. doi: 10.1016/j.it.2022.04.010
2. Rezaei M, Tan J, Zeng C, Li Y, Ganjalikhani-Hakemi M. TIM-3 in leukemia; immune response and beyond. Front Oncol. (2021) 11:753677. doi: 10.3389/fonc.2021.753677
3. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
4. Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The PD-1/PD-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell. (2020) 38:685–700. doi: 10.1016/j.ccell.2020.09.001
5. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. (2018) 128:4654–68. doi: 10.1172/JCI99317
6. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discovery. (2019) 9:1422–37. doi: 10.1158/2159-8290.CD-18-1259
7. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. (2006) 313:1960–64. doi: 10.1126/science.1129139
8. Galon J, Fridman WH, Pages F. The adaptive immunologic microenvironment in colorectal cancer: a novel perspective. Cancer Res. (2007) 67:1883–86. doi: 10.1158/0008-5472.CAN-06-4806
9. Camus M, Tosolini M, Trajanoski Z, Herman Fridman W, Galon J, Mlecnik B, et al. Coordination of intratumoral immune reaction and human colorectal cancer recurrence. Cancer Res (Chicago Ill.). (2009) 69:2685–93. doi: 10.1158/0008-5472.CAN-08-2654
10. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of Malignant tumours. J Pathol. (2014) 232:199–209. doi: 10.1002/path.4287
11. Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. (2018) 391:2128–39. doi: 10.1016/S0140-6736(18)30789-X
12. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
13. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. (2019) 19:665–74. doi: 10.1038/s41577-019-0221-9
14. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. (2017) 545:452–56. doi: 10.1038/nature22367
15. Johnson DB, Nebhan CA, Moslehi JJ, Balko JM. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol. (2022) 19:254–67. doi: 10.1038/s41571-022-00600-w
16. Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. (2021) 11:5365–86. doi: 10.7150/thno.58390
17. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. (2014) 14:135–46. doi: 10.1038/nrc3670
18. Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. (2019) 25:767–75. doi: 10.1038/s41591-019-0434-2
19. Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure JP, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. (2018) 10:767–75. doi: 10.1126/scitranslmed.aau5516
20. Kahles A, Lehmann KV, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. (2018) 34:211–24. doi: 10.1016/j.ccell.2018.07.001
21. Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, et al. Microsatellite instability is associated with the presence of lynch syndrome pan-cancer. J Clin Oncol. (2019) 37:286–95. doi: 10.1200/JCO.18.00283
22. Fan A, Wang B, Wang X, Nie Y, Fan D, Zhao X, et al. Immunotherapy in colorectal cancer: current achievements and future perspective. Int J Biol Sci. (2021) 17:3837–49. doi: 10.7150/ijbs.64077
23. Jardim DL, Goodman A, de Melo GD, Kurzrock R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell. (2021) 39:154–73. doi: 10.1016/j.ccell.2020.10.001
24. McGrail DJ, Federico L, Li Y, Dai H, Lu Y, Mills GB, et al. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat Commun. (2018) 9:1317. doi: 10.1038/s41467-018-03730-x
25. Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U.S.A. (2016) 113:E7759–68. doi: 10.1073/pnas.1609376113
26. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
27. de Mingo PÁ, Hänggi K, Celias DP, Gardner A, Li J, Batista-Bittencourt B, et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity. (2021) 54:1154–67. doi: 10.1016/j.immuni.2021.04.019
28. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. (2008) 322:1097–100. doi: 10.1126/science.1164206
29. Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martínez-Cano S, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. (2017) 8:16073. doi: 10.1038/ncomms16073
30. Feng M, Jiang W, Kim B, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. (2019) 19:568–86. doi: 10.1038/s41568-019-0183-z
31. Luo M, Wang X, Wu S, Yang C, Su Q, Huang L, et al. A20 promotes colorectal cancer immune evasion by upregulating STC1 expression to block “eat-me” signal. Signal Transduct Target Ther. (2023) 8:312. doi: 10.1038/s41392-023-01545-x
32. Lin H, Kryczek I, Li S, Green MD, Ali A, Hamasha R, et al. Stanniocalcin 1 is a phagocytosis checkpoint driving tumor immune resistance. Cancer Cell. (2021) 39:480–93. doi: 10.1016/j.ccell.2020.12.023
33. Song X, Zhou Z, Li H, Xue Y, Lu X, Bahar I, et al. Pharmacologic suppression of B7-H4 glycosylation restores antitumor immunity in immune-cold breast cancers. Cancer Discovery. (2020) 10:1872–93. doi: 10.1158/2159-8290.CD-20-0402
34. Giampazolias E, Schulz O, Lim K, Rogers NC, Chakravarty P, Srinivasan N, et al. Secreted gelsolin inhibits DNGR-1-dependent cross-presentation and cancer immunity. Cell. (2021) 184:4016–31. doi: 10.1016/j.cell.2021.05.021
35. Montesion M, Murugesan K, Jin DX, Sharaf R, Sanchez N, Guria A, et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discovery. (2021) 11:282–92. doi: 10.1158/2159-8290.CD-20-0672
36. Chowell D, Morris L, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. (2018) 359:582–87. doi: 10.1126/science.aao4572
37. Gu SS, Zhang W, Wang X, Jiang P, Traugh N, Li Z, et al. Therapeutically increasing MHC-I expression potentiates immune checkpoint blockade. Cancer Discovery. (2021) 11:1524–41. doi: 10.1158/2159-8290.CD-20-0812
38. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. (2020) 581:100–05. doi: 10.1038/s41586-020-2229-5
39. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
40. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. (2015) 527:249–53. doi: 10.1038/nature15520
41. Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 epigenetically silences th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. (2016) 76:275–82. doi: 10.1158/0008-5472.CAN-15-1938
42. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell. (2019) 35:885–900. doi: 10.1016/j.ccell.2019.05.004
43. Molon B, Ugel S, Del PF, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
44. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–48. doi: 10.1038/nature25501
45. Tauriello D, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
46. Vinokurova D, Apetoh L. The emerging role of IL-9 in the anticancer effects of anti-PD-1 therapy. Biomolecules. (2023) 13:5225–37. doi: 10.3390/biom13040670
47. Falcomatà C, Bärthel S, Schneider G, Rad R, Schmidt-Supprian M, Saur D. Context-specific determinants of the immunosuppressive tumor microenvironment in pancreatic cancer. Cancer Discovery. (2023) 13:278–97. doi: 10.1158/2159-8290.CD-22-0876
48. Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. (2022) 15:61. doi: 10.1186/s13045-022-01282-8
49. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64. doi: 10.4049/jimmunol.155.3.1151
50. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. (2001) 167:1245–53. doi: 10.4049/jimmunol.167.3.1245
51. Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med. (2001) 193:1285–94. doi: 10.1084/jem.193.11.1285
52. Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med. (2001) 193:1303–10. doi: 10.1084/jem.193.11.1303
53. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
54. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–36. doi: 10.1038/ni904
55. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. (2003) 4:337–42. doi: 10.1038/ni909
56. Lee W, Lee GR. Transcriptional regulation and development of regulatory T cells. Exp Mol Med. (2018) 50:e456. doi: 10.1038/emm.2017.313
57. Sharma A, Rudra D. Emerging functions of regulatory T cells in tissue homeostasis. Front Immunol. (2018) 9:883. doi: 10.3389/fimmu.2018.00883
58. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
59. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
60. Itahashi K, Irie T, Nishikawa H. Regulatory T-cell development in the tumor microenvironment. Eur J Immunol. (2022) 52:1216–27. doi: 10.1002/eji.202149358
61. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. (2019) 110:2080–89. doi: 10.1111/cas.14069
62. Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U.S.A. (2005) 102:419–24. doi: 10.1073/pnas.0408197102
63. Yano H, Andrews LP, Workman CJ, Vignali D. Intratumoral regulatory T cells: markers, subsets and their impact on anti-tumor immunity. Immunology. (2019) 157:232–47. doi: 10.1111/imm.13067
64. Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. (2023) 41:450–65. doi: 10.1016/j.ccell.2023.02.014
65. DeLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. (2012) 18:3022–29. doi: 10.1158/1078-0432.CCR-11-3216
66. Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, et al. A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res. (2015) 75:3800–11. doi: 10.1158/0008-5472.CAN-15-1082
67. Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol. (2013) 91:493–502. doi: 10.1038/icb.2013.29
68. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. (2014) 63:247–57. doi: 10.1007/s00262-013-1508-5
69. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
70. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U.S.A. (2014) 111:11774–79. doi: 10.1073/pnas.1410626111
71. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. (2016) 539:443–47. doi: 10.1038/nature20554
72. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. (2015) 66:97–110. doi: 10.1146/annurev-med-051013-052304
73. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). (2014) 6:1670–90. doi: 10.3390/cancers6031670
74. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity. (2016) 44:303–15. doi: 10.1016/j.immuni.2016.01.014
75. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
76. Madsen DH, Leonard D, Masedunskas A, Moyer A, Jürgensen HJ, Peters DE, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol. (2013) 202:951–66. doi: 10.1083/jcb.201301081
77. Kawachi A, Yoshida H, Kitano S, Ino Y, Kato T, Hiraoka N. Tumor-associated CD204(+) M2 macrophages are unfavorable prognostic indicators in uterine cervical adenocarcinoma. Cancer Sci. (2018) 109:863–70. doi: 10.1111/cas.13476
78. Hu W, Li X, Zhang C, Yang Y, Jiang J, Wu C. Tumor-associated macrophages in cancers. Clin Transl Oncol. (2016) 18:251–58. doi: 10.1007/s12094-015-1373-0
79. Fritz JM, Tennis MA, Orlicky DJ, Lin H, Ju C, Redente EF, et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol. (2014) 5:587. doi: 10.3389/fimmu.2014.00587
80. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
81. Georganaki M, van Hooren L, Dimberg A. Vascular targeting to increase the efficiency of immune checkpoint blockade in cancer. Front Immunol. (2018) 9:3081. doi: 10.3389/fimmu.2018.03081
82. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. (2008) 14:28–36. doi: 10.1038/nm1699
83. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. (2019) 176:1248–64. doi: 10.1016/j.cell.2019.01.021
84. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
85. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. (2016) 22:851–60. doi: 10.1038/nm.4123
86. Huang Y, Kim B, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat Rev Immunol. (2018) 18:195–203. doi: 10.1038/nri.2017.145
87. McDonald PC, Chafe SC, Dedhar S. Overcoming hypoxia-mediated tumor progression: combinatorial approaches targeting pH regulation, angiogenesis and immune dysfunction. Front Cell Dev Biol. (2016) 4:27. doi: 10.3389/fcell.2016.00027
88. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
89. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. (2020) 17:611–29. doi: 10.1038/s41571-020-0382-2
90. Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. (2013) 13:842–57. doi: 10.1038/nrc3613
91. Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein kinase C and cancer: what we know and what we do not. Oncogene. (2014) 33:5225–37. doi: 10.1038/onc.2013.524
92. Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discovery. (2012) 11:937–57. doi: 10.1038/nrd3871
93. Kikkawa U, Takai Y, Tanaka Y, Miyake R, Nishizuka Y. Protein kinase C as a possible receptor protein of tumor-promoting phorbol esters. J Biol Chem. (1983) 258:11442–45. doi: 10.1016/S0021-9258(17)44245-1
94. Sadeghi MM, Salama MF, Hannun YA. Protein kinase C as a therapeutic target in non-small cell lung cancer. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22115527
95. Black AR, Black JD. Protein kinase C signaling and cell cycle regulation. Front Immunol. (2012) 3:423. doi: 10.3389/fimmu.2012.00423
96. Cheng Y, Zhu Y, Xu W, Xu J, Yang M, Chen P, et al. PKCα in colon cancer cells promotes M1 macrophage polarization via MKK3/6-P38 MAPK pathway. Mol Carcinog. (2018) 57:1017–29. doi: 10.1002/mc.22822
97. Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, et al. Protein kinase C theta affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med. (2003) 197:1525–35. doi: 10.1084/jem.20020234
98. Kwon MJ, Ma J, Ding Y, Wang R, Sun Z. Protein kinase C-θ promotes Th17 differentiation via upregulation of Stat3. J Immunol. (2012) 188:5887–97. doi: 10.4049/jimmunol.1102941
99. He X, Koenen H, Smeets RL, Keijsers R, van Rijssen E, Koerber A, et al. Targeting PKC in human T cells using sotrastaurin (AEB071) preserves regulatory T cells and prevents IL-17 production. J Invest Dermatol. (2014) 134:975–83. doi: 10.1038/jid.2013.459
100. Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS, et al. Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J Clin Invest. (1996) 98:2018–26. doi: 10.1172/JCI119006
101. Suzuma K, Takahara N, Suzuma I, Isshiki K, Ueki K, Leitges M, et al. Characterization of protein kinase C beta isoform’s action on retinoblastoma protein phosphorylation, vascular endothelial growth factor-induced endothelial cell proliferation, and retinal neovascularization. Proc Natl Acad Sci U.S.A. (2002) 99:721–26. doi: 10.1073/pnas.022644499
102. Goicoechea SM, García-Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG, et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene. (2014) 33:1265–73. doi: 10.1038/onc.2013.68
103. Hennings H, Blumberg PM, Pettit GR, Herald CL, Shores R, Yuspa SH. Bryostatin 1, an activator of protein kinase C, inhibits tumor promotion by phorbol esters in SENCAR mouse skin. Carcinogenesis. (1987) 8:1343–46. doi: 10.1093/carcin/8.9.1343
104. Boyle GM, D’Souza MM, Pierce CJ, Adams RA, Cantor AS, Johns JP, et al. Intra-lesional injection of the novel PKC activator EBC-46 rapidly ablates tumors in mouse models. PloS One. (2014) 9:e108887. doi: 10.1371/journal.pone.0108887
105. Miller J, Campbell J, Blum A, Reddell P, Gordon V, Schmidt P, et al. Dose characterization of the investigational anticancer drug tigilanol tiglate (EBC-46) in the local treatment of canine mast cell tumors. Front Vet Sci. (2019) 6:106. doi: 10.3389/fvets.2019.00106
106. De Ridder TR, Campbell JE, Burke-Schwarz C, Clegg D, Elliot EL, Geller S, et al. Randomized controlled clinical study evaluating the efficacy and safety of intratumoral treatment of canine mast cell tumors with tigilanol tiglate (EBC-46). J Vet Intern Med. (2021) 35:415–29. doi: 10.1111/jvim.15806
107. Utz I, Hofer S, Regenass U, Hilbe W, Thaler J, Grunicke H, et al. The protein kinase C inhibitor CGP 41251, a staurosporine derivative with antitumor activity, reverses multidrug resistance. Int J Cancer. (1994) 57:104–10. doi: 10.1002/ijc.2910570119
108. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. (2012) 13:283–96. doi: 10.1038/nrm3330
109. Wang Z, Wu X. Study and analysis of antitumor resistance mechanism of PD1/PD-L1 immune checkpoint blocker. Cancer Med. (2020) 9:8086–121. doi: 10.1002/cam4.3410
110. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. (2017) 9:104–10. doi: 10.1126/scitranslmed.aah3560
111. Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. (2011) 19:629–39. doi: 10.1016/j.ccr.2011.03.023
112. Liu JJ, Li Y, Chen WS, Liang Y, Wang G, Zong M, et al. Shp2 deletion in hepatocytes suppresses hepatocarcinogenesis driven by oncogenic β-Catenin, PIK3CA and MET. J Hepatol. (2018) 69:79–88. doi: 10.1016/j.jhep.2018.02.014
113. Song M, Chen D, Lu B, Wang C, Zhang J, Huang L, et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PloS One. (2013) 8:e65821. doi: 10.1371/journal.pone.0065821
114. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
115. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discovery. (2016) 6:202–16. doi: 10.1158/2159-8290.CD-15-0283
116. de Gramont A, Faivre S, Raymond E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology. (2017) 6:e1257453. doi: 10.1080/2162402X.2016.1257453
117. Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux I, Bajard A, et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. (2011) 71:5423–34. doi: 10.1158/0008-5472.CAN-11-0367
118. Sisirak V, Faget J, Vey N, Blay JY, Ménétrier-Caux C, Caux C, et al. Plasmacytoid dendritic cells deficient in IFNα production promote the amplification of FOXP3(+) regulatory T cells and are associated with poor prognosis in breast cancer patients. Oncoimmunology. (2013) 2:e22338. doi: 10.4161/onci.22338
119. Yang L, Moses HL. Transforming growth factor beta: tumor suppressor or promoter? Are host immune cells the answer? Cancer Res. (2008) 68:9107–11. doi: 10.1158/0008-5472.CAN-08-2556
120. Batlle E, Massagué J. Transforming growth factor-β Signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
121. Derynck R, Turley SJ, Akhurst RJ. TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. (2021) 18:9–34. doi: 10.1038/s41571-020-0403-1
122. Xie F, Ling L, van Dam H, Zhou F, Zhang L. TGF-β signaling in cancer metastasis. Acta Biochim Biophys Sin (Shanghai). (2018) 50:121–32. doi: 10.1093/abbs/gmx123
123. An X, Zhu Y, Zheng T, Wang G, Zhang M, Li J, et al. An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol Ther Nucleic Acids. (2019) 14:80–9. doi: 10.1016/j.omtn.2018.11.003
124. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. (2018) 553:467–72. doi: 10.1038/nature25432
125. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
126. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
127. Li J, Duran MA, Dhanota N, Chatila WK, Bettigole SE, Kwon J, et al. Metastasis and immune evasion from extracellular cGAMP hydrolysis. Cancer Discov. (2021) 11:1212–27. doi: 10.1158/2159-8290.CD-20-0387
128. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. (2016) 533:493–98. doi: 10.1038/nature18268
129. Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat Rev Immunol. (2003) 3:544–56. doi: 10.1038/nri1131
130. Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. (2010) 33:326–39. doi: 10.1016/j.immuni.2010.09.006
131. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–47. doi: 10.1016/1074-7613(95)90125-6
132. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. (1995) 270:985–88. doi: 10.1126/science.270.5238.985
133. Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. (2002) 2:116–26. doi: 10.1038/nri727
134. Wu K, Yi M, Qin S, Chu Q, Zheng X, Wu K. The efficacy and safety of combination of PD-1 and CTLA-4 inhibitors: a meta-analysis. Exp Hematol Oncol. (2019) 8:26. doi: 10.1186/s40164-019-0150-0
135. Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. (2016) 17:1558–68. doi: 10.1016/S1470-2045(16)30366-7
136. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2019) 381:1535–46. doi: 10.1056/NEJMoa1910836
137. Overman MJ, Lonardi S, Wong K, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. (2018) 36:773–79. doi: 10.1200/JCO.2017.76.9901
138. Motzer RJ, Rini BI, McDermott DF, Arén FO, Hammers HJ, Carducci MA, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. (2019) 20:1370–85. doi: 10.1016/S1470-2045(19)30413-9
139. Hellmann MD, Paz-Ares L, Bernabe CR, Zurawski B, Kim SW, Carcereny CE, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
140. Baas P, Scherpereel A, Nowak AK, Fujimoto N, Peters S, Tsao AS, et al. First-line nivolumab plus ipilimumab in unresectable Malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. (2021) 397:375–86. doi: 10.1016/S0140-6736(20)32714-8
141. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
142. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
143. Harding JJ, Moreno V, Bang YJ, Hong MH, Patnaik A, Trigo J, et al. Blocking TIM-3 in treatment-refractory advanced solid tumors: A phase ia/b study of LY3321367 with or without an anti-PD-L1 antibody. Clin Cancer Res. (2021) 27:2168–78. doi: 10.1158/1078-0432.CCR-20-4405
144. Hollebecque A, Chung HC, de Miguel MJ, Italiano A, Machiels JP, Lin CC, et al. Safety and antitumor activity of α-PD-L1 antibody as monotherapy or in combination with α-TIM-3 antibody in patients with microsatellite instability-high/mismatch repair-deficient tumors. Clin Cancer Res. (2021) 27:6393–404. doi: 10.1158/1078-0432.CCR-21-0261
145. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res. (2021) 27:3620–29. doi: 10.1158/1078-0432.CCR-20-4746
146. Gestermann N, Saugy D, Martignier C, Tillé L, Fuertes MS, Zettl M, et al. LAG-3 and PD-1+LAG-3 inhibition promote anti-tumor immune responses in human autologous melanoma/T cell co-cultures. Oncoimmunology. (2020) 9:1736792. doi: 10.1080/2162402X.2020.1736792
147. Li Y, Zhang Y, Cao G, Zheng X, Sun C, Wei H, et al. Blockade of checkpoint receptor PVRIG unleashes anti-tumor immunity of NK cells in murine and human solid tumors. J Hematol Oncol. (2021) 14:100. doi: 10.1186/s13045-021-01112-3
148. Mao L, Xiao Y, Yang QC, Yang SC, Yang LL, Sun ZJ. TIGIT/CD155 blockade enhances anti-PD-L1 therapy in head and neck squamous cell carcinoma by targeting myeloid-derived suppressor cells. Oral Oncol. (2021) 121:105472. doi: 10.1016/j.oraloncology.2021.105472
149. Xiao N, Zhu X, Li K, Chen Y, Liu X, Xu B, et al. Blocking siglec-10(hi) tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp Hematol Oncol. (2021) 10:36. doi: 10.1186/s40164-021-00230-5
150. Lipson EJ, Tawbi HA, SChadendorf D, Ascierto PA, Matamala L, Gutiérrez EC, et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J Clin Oncol. (2021) 39:9503. doi: 10.1200/JCO.2021.39.15_suppl.9503
151. Vaena DA, Fleming GF, Chmielowski B, Sharma M, Hamilton EP, Sullivan RJ, et al. COM701 with or without nivolumab: Results of an ongoing phase 1 study of safety, tolerability and preliminary antitumor activity in patients with advanced solid Malignancies (NCT03667716). J Clin Oncol. (2021) 39:2504. doi: 10.1200/JCO.2021.39.15_suppl.2504
152. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J Clin Oncol. (2020) 38:9503. doi: 10.1200/JCO.2020.38.15_suppl.9503
153. Daei SA, Mohamed KL, Sarkesh A, Mardi A, Aghebati-Maleki A, Aghebati-Maleki L, et al. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front Immunol. (2023) 14:1113882. doi: 10.3389/fimmu.2023.1113882
154. Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric antigen receptors T cell therapy in solid tumor: challenges and clinical applications. Front Immunol. (2017) 8:1850. doi: 10.3389/fimmu.2017.01850
155. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
156. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
157. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105:3051–57. doi: 10.1182/blood-2004-07-2974
158. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. (2002) 295:2097–100. doi: 10.1126/science.1068440
159. Simonetta F, Alvarez M, Negrin RS. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front Immunol. (2017) 8:465. doi: 10.3389/fimmu.2017.00465
160. Huang RS, Lai MC, Shih HA, Lin S. A robust platform for expansion and genome editing of primary human natural killer cells. J Exp Med. (2021) 218:5636–46. doi: 10.1084/jem.20201529
161. Suen WC, Lee WY, Leung KT, Pan XH, Li G. Natural killer cell-based cancer immunotherapy: A review on 10 years completed clinical trials. Cancer Invest. (2018) 36:431–57. doi: 10.1080/07357907.2018.1515315
162. Wang W, Jiang J, Wu C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. (2020) 472:175–80. doi: 10.1016/j.canlet.2019.11.033
163. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92. doi: 10.1016/j.stem.2018.06.002
164. Quintarelli C, Sivori S, Caruso S, Carlomagno S, Falco M, Boffa I, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia. (2020) 34:1102–15. doi: 10.1038/s41375-019-0613-7
165. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
166. Zhang L, Meng Y, Feng X, Han Z. CAR-NK cells for cancer immunotherapy: from bench to bedside. biomark Res. (2022) 10:12. doi: 10.1186/s40364-022-00364-6
167. van de Donk N, Zweegman S. T-cell-engaging bispecific antibodies in cancer. Lancet. (2023) 402:142–58. doi: 10.1016/S0140-6736(23)00521-4
168. Wei J, Yang Y, Wang G, Liu M. Current landscape and future directions of bispecific antibodies in cancer immunotherapy. Front Immunol. (2022) 13:6537. doi: 10.3389/fimmu.2022.1035276
169. Rezaei M, Danilova ND, Soltani M, Savvateeva LV, Tarasov VV, Ganjalikhani-Hakemi M, et al. Cancer vaccine in cold tumors: clinical landscape, challenges, and opportunities. Curr Cancer Drug Targets. (2022) 22:437–53. doi: 10.2174/1568009622666220214103533
170. Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. (2021) 18:215–29. doi: 10.1038/s41571-020-00460-2
171. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
172. Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight. (2018) 3:6537. doi: 10.1172/jci.insight.122857
173. Zhu G, Lynn GM, Jacobson O, Chen K, Liu Y, Zhang H, et al. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat Commun. (2017) 8:1954. doi: 10.1038/s41467-017-02191-y
174. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. (2017) 547:222–26. doi: 10.1038/nature23003
175. Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, et al. A phase ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell. (2020) 183:347–62. doi: 10.1016/j.cell.2020.08.053
176. Innao V, Rizzo V, Allegra AG, Musolino C, Allegra A. Oncolytic viruses and hematological Malignancies: A new class of immunotherapy drugs. Curr Oncol. (2020) 28:159–83. doi: 10.3390/curroncol28010019
177. Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. (2018) 36:1658–67. doi: 10.1200/JCO.2017.73.7379
178. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. (2018) 10:222–6. doi: 10.1126/scitranslmed.aam7577
179. Bourgeois-Daigneault MC, Roy DG, Aitken AS, El SN, Martin NT, Varette O, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. (2018) 10:362–47. doi: 10.1126/scitranslmed.aao1641
180. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
181. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. (2021) 6:75. doi: 10.1038/s41392-021-00484-9
182. Harada K, Dong X, Estrella JS, Correa AM, Xu Y, Hofstetter WL, et al. Tumor-associated macrophage infiltration is highly associated with PD-L1 expression in gastric adenocarcinoma. Gastric Cancer. (2018) 21:31–40. doi: 10.1007/s10120-017-0760-3
183. Tsukamoto M, Imai K, Ishimoto T, Komohara Y, Yamashita YI, Nakagawa S, et al. PD-L1 expression enhancement by infiltrating macrophage-derived tumor necrosis factor-α leads to poor pancreatic cancer prognosis. Cancer Sci. (2019) 110:310–20. doi: 10.1111/cas.13874
184. Omstead AN, Paskewicz M, Gorbunova A, Zheng P, Salvitti MS, Mansoor R, et al. CSF-1R inhibitor, pexidartinib, sensitizes esophageal adenocarcinoma to PD-1 immune checkpoint blockade in a rat model. Carcinogenesis. (2022) 43:842–50. doi: 10.1093/carcin/bgac043
185. Shi G, Yang Q, Zhang Y, Jiang Q, Lin Y, Yang S, et al. Modulating the tumor microenvironment via oncolytic viruses and CSF-1R inhibition synergistically enhances anti-PD-1 immunotherapy. Mol Ther. (2019) 27:244–60. doi: 10.1016/j.ymthe.2018.11.010
186. Gomez-Roca C, Cassier P, Zamarin D, Machiels JP, Perez GJ, Stephen HF, et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naïve or experienced for immune checkpoint blockade. J Immunother Cancer. (2022) 10:799–820. doi: 10.1136/jitc-2021-004076
187. Haag GM, Springfeld C, Grün B, Apostolidis L, Zschäbitz S, Dietrich M, et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer - The PICCASSO phase I trial. Eur J Cancer. (2022) 167:112–22. doi: 10.1016/j.ejca.2022.03.017
188. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. (2006) 203:1259–71. doi: 10.1084/jem.20052494
189. Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. (2008) 15:3–12. doi: 10.1038/sj.cdd.4402269
190. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. (2014) 124:687–95. doi: 10.1172/JCI67313
191. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. (2015) 15:409–25. doi: 10.1038/nrc3958
192. Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. (2018) 18:313–22. doi: 10.1038/nrc.2018.6
193. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. (2012) 366:925–31. doi: 10.1056/NEJMoa1112824
194. Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. (2016) 40:25–37. doi: 10.1016/j.currproblcancer.2015.10.001
195. Park SS, Dong H, Liu X, Harrington SM, Krco CJ, Grams MP, et al. PD-1 restrains radiotherapy-induced abscopal effect. Cancer Immunol Res. (2015) 3:610–19. doi: 10.1158/2326-6066.CIR-14-0138
196. Reynders K, Illidge T, Siva S, Chang JY, De Ruysscher D. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer Treat Rev. (2015) 41:503–10. doi: 10.1016/j.ctrv.2015.03.011
197. Dudzinski SO, Cameron BD, Wang J, Rathmell JC, Giorgio TD, Kirschner AN. Combination immunotherapy and radiotherapy causes an abscopal treatment response in a mouse model of castration resistant prostate cancer. J Immunother Cancer. (2019) 7:218. doi: 10.1186/s40425-019-0704-z
198. Ji D, Song C, Li Y, Xia J, Wu Y, Jia J, et al. Combination of radiotherapy and suppression of Tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J Immunother Cancer. (2020) 8:313–22. doi: 10.1136/jitc-2020-000826
199. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. (2018) 379:2342–50. doi: 10.1056/NEJMoa1809697
200. Fizazi K, Drake CG, Beer TM, Kwon ED, Scher HI, Gerritsen WR, et al. Final analysis of the ipilimumab versus placebo following radiotherapy phase III trial in postdocetaxel metastatic castration-resistant prostate cancer identifies an excess of long-term survivors. Eur Urol. (2020) 78:822–30. doi: 10.1016/j.eururo.2020.07.032
201. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. (2013) 39:74–88. doi: 10.1016/j.immuni.2013.06.014
202. Wang Q, Ju X, Wang J, Fan Y, Ren M, Zhang H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett. (2018) 438:17–23. doi: 10.1016/j.canlet.2018.08.028
203. Rodríguez-Abreu D, Powell SF, Hochmair MJ, Gadgeel S, Esteban E, Felip E, et al. Pemetrexed plus platinum with or without pembrolizumab in patients with previously untreated metastatic nonsquamous NSCLC: protocol-specified final analysis from KEYNOTE-189. Ann Oncol. (2021) 32:881–95. doi: 10.1016/j.annonc.2021.04.008
204. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. (2020) 396:1817–28. doi: 10.1016/S0140-6736(20)32531-9
205. Awad MM, Gadgeel SM, Borghaei H, Patnaik A, Yang JC, Powell SF, et al. Long-term overall survival from KEYNOTE-021 cohort G: pemetrexed and carboplatin with or without pembrolizumab as first-line therapy for advanced nonsquamous NSCLC. J Thorac Oncol. (2021) 16:162–68. doi: 10.1016/j.jtho.2020.09.015
206. Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. (2018) 379:2220–29. doi: 10.1056/NEJMoa1809064
207. Ni JJ, Zhang ZZ, Ge MJ, Chen JY, Zhuo W. Immune-based combination therapy to convert immunologically cold tumors into hot tumors: an update and new insights. Acta Pharmacol Sin. (2023) 44:288–307. doi: 10.1038/s41401-022-00953-z
208. Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2020) 395:1835–44. doi: 10.1016/S0140-6736(20)30934-X
209. Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. (2020) 21:1155–64. doi: 10.1016/S1470-2045(20)30324-7
210. Khoo LT, Chen LY. Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep. (2018) 19:1817–28. doi: 10.15252/embr.201846935
211. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
212. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature. (2013) 498:380–84. doi: 10.1038/nature12306
213. Jiang M, Chen P, Wang L, Li W, Chen B, Liu Y, et al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol. (2020) 13:81. doi: 10.1186/s13045-020-00916-z
214. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. (2014) 88:5328–41. doi: 10.1128/JVI.00037-14
215. Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. (2013) 34:67–73. doi: 10.1016/j.it.2012.10.004
216. Lara PJ, Douillard JY, Nakagawa K, von Pawel J, McKeage MJ, Albert I, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. (2011) 29:2965–71. doi: 10.1200/JCO.2011.35.0660
217. Shih AY, Damm-Ganamet KL, Mirzadegan T. Dynamic structural differences between human and mouse STING lead to differing sensitivity to DMXAA. Biophys J. (2018) 114:32–9. doi: 10.1016/j.bpj.2017.10.027
218. Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. (2013) 190:5216–25. doi: 10.4049/jimmunol.1300097
219. Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. (2013) 14:19–26. doi: 10.1038/ni.2491
220. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. (2011) 478:515–18. doi: 10.1038/nature10429
221. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. (2015) 11:1018–30. doi: 10.1016/j.celrep.2015.04.031
222. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. (2018) 564:439–43. doi: 10.1038/s41586-018-0705-y
223. Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An orally available non-nucleotide STING agonist with antitumor activity. Science. (2020) 369:32–9. doi: 10.1126/science.aba6098
224. Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. (2020) 30:966–79. doi: 10.1038/s41422-020-00395-4
225. Wang C, Guan Y, Lv M, Zhang R, Guo Z, Wei X, et al. Manganese Increases the Sensitivity of the cGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity. (2018) 48:675–87. doi: 10.1016/j.immuni.2018.03.017
226. Nakamura T, Sato T, Endo R, Sasaki S, Takahashi N, Sato Y, et al. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J Immunother Cancer. (2021) 9:515–8. doi: 10.1136/jitc-2021-002852
227. Yi M, Niu M, Zhang J, Li S, Zhu S, Yan Y, et al. Combine and conquer: manganese synergizing anti-TGF-β/PD-L1 bispecific antibody YM101 to overcome immunotherapy resistance in non-inflamed cancers. J Hematol Oncol. (2021) 14:146. doi: 10.1186/s13045-021-01155-6
228. Song Y, Liu Y, Teo HY, Hanafi ZB, Mei Y, Zhu Y, et al. Manganese enhances the antitumor function of CD8(+) T cells by inducing type I interferon production. Cell Mol Immunol. (2021) 18:1571–74. doi: 10.1038/s41423-020-00524-4
229. Meric-Bernstam F, Sandhu SK, Hamid O, Spreafico A, Kasper S, Dummer R, et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J Clin Oncol. (2019) 37:2507. doi: 10.1200/JCO.2019.37.15_suppl.2507
230. Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. (2018) 29:viii712. doi: 10.1093/annonc/mdy424.015
231. Rodallec A, Sicard G, Fanciullino R, Benzekry S, Lacarelle B, Milano G, et al. Turning cold tumors into hot tumors: harnessing the potential of tumor immunity using nanoparticles. Expert Opin Drug Metab Toxicol. (2018) 14:1139–47. doi: 10.1080/17425255.2018.1540588
232. Duan X, Chan C, Lin W. Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew Chem Int Ed Engl. (2019) 58:670–80. doi: 10.1002/anie.201804882
233. Song P, Han X, Zheng R, Yan J, Wu X, Wang Y, et al. Upregulation of MHC-I and downregulation of PD-L1 expression by doxorubicin and deferasirox codelivered liposomal nanoparticles for chemoimmunotherapy of melanoma. Int J Pharm. (2022) 624:122002. doi: 10.1016/j.ijpharm.2022.122002
Keywords: cold tumors, immune checkpoint inhibitors, tumor-infiltrating T lymphocytes, tumor microenvironment, immunotherapy
Citation: Ouyang P, Wang L, Wu J, Tian Y, Chen C, Li D, Yao Z, Chen R, Xiang G, Gong J and Bao Z (2024) Overcoming cold tumors: a combination strategy of immune checkpoint inhibitors. Front. Immunol. 15:1344272. doi: 10.3389/fimmu.2024.1344272
Received: 25 November 2023; Accepted: 26 February 2024;
Published: 13 March 2024.
Edited by:
Mazdak Ganjalikhani Hakemi, Isfahan University of Medical Sciences, IranReviewed by:
Muzamil Y. Want, Children’s Hospital of Philadelphia, United StatesVida Homayouni, Isfahan University of Medical Sciences, Iran
Copyright © 2024 Ouyang, Wang, Wu, Tian, Chen, Li, Yao, Chen, Xiang, Gong and Bao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoan Xiang, guoanx_fimmu_edu@163.com; Jin Gong, zaitushuguan@163.com; Zhen Bao, baozhenjnu@126.com
†These authors have contributed equally to this work